
CO/Pd (100)的结构优化
版本:2015.1
在本教程中,您将学习如何用 Fixed 和 Rigid 约束原子并进行结构优化。学习案例为 CO 分子在 Pd(100) 面上的吸附,所选计算工具为 ATK-DFT 计算引擎和标准 GGA 泛函。
具体地,您将:
- 优化块体钯晶体;
- 切割块体获得 Pd(100) 面,然后弛豫结构;
- 表面吸附 CO 分子,按以下 2 步对体系进行弛豫;
- 初始弛豫期间,给原子施加 Rigid 约束;
- 最终优化时,对底部的 Pd 原子施加 Fixed 约束。
- 最后,弛豫 CO 分子,计算吸附能。
提示
本教程使用特定版本的QuantumATK创建,因此涉及的截图和脚本参数可能与您实际使用的版本略有区别,请在学习时务必注意。

钯块体
创建一个新的项目,命名为 “Pd100_CO”,打开 Builder ![]() 。
。
- 点击 Add
 From Database 将钯块体结构导入到 Stash,然后用发送按钮
From Database 将钯块体结构导入到 Stash,然后用发送按钮  传送构形到 Script Generator
传送构形到 Script Generator  。
。 - 在 Script Generator 中,双击添加
 New Claculator 和
New Claculator 和  OptimizeGeometry 模块到脚本,将默认输出文件名称改为
OptimizeGeometry 模块到脚本,将默认输出文件名称改为 Pd_bulk.nc。
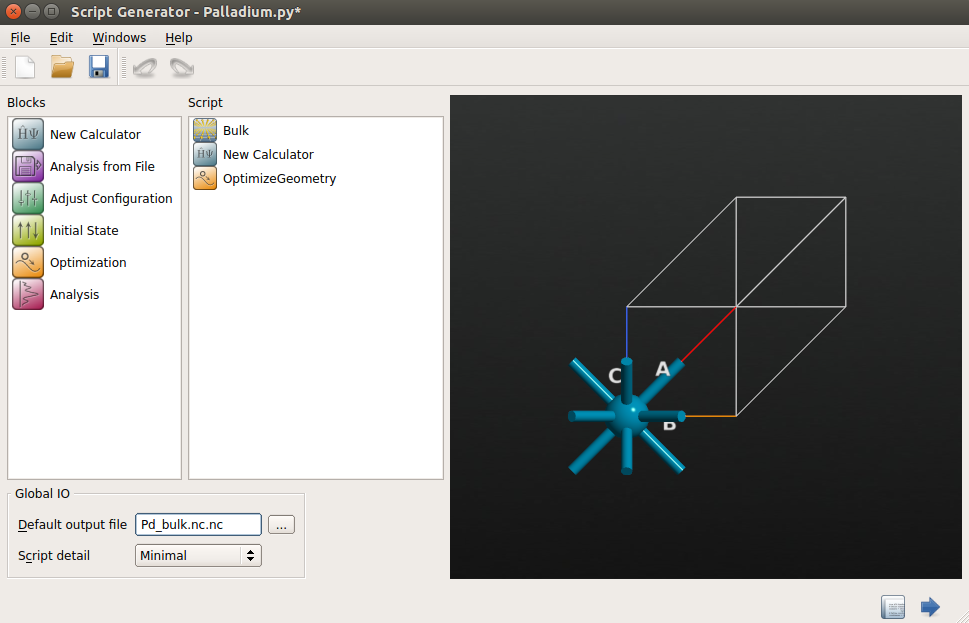
- 双击 New Claculator 模块,在随即打开的窗口中设置参数:
- k-point sampling:9×9×9;
- exchange-correlation:GGA-PBE 泛函。
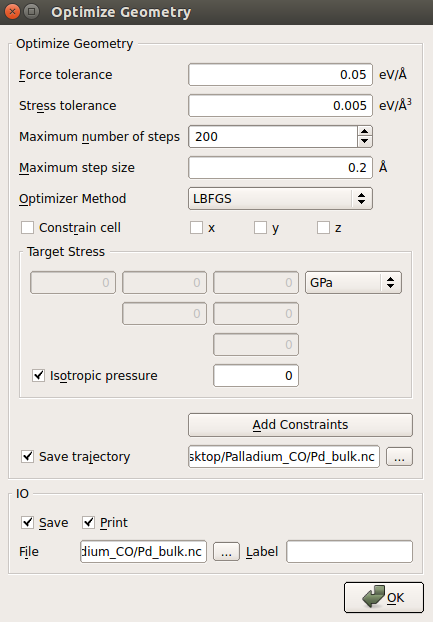
- 打开 OptimizeGeometry 模块,如下图所示设置参数。特别地:
- 不勾选 Constrain cell 以确保应力弛豫;
- 降低 Stress tolerance 至 0.005 eV/Å3;
- 点击 Save trajectory 保存弛豫的轨迹,输入轨迹文件名称为
Pd_bulk.nc。
提示
通常,保存轨迹文件是一个很好的做法。假如您的结构优化过程中断,可以轻易地从轨迹的最后一步重启计算。如果您想知道如何重启中断的计算,可以参考教程 Restarting stopped calculations。
- 单击 OK,结构优化窗口关闭。
块体弛豫结束后,计算的输出结果会在 LabFloor 显示。ID 名字含有 gID000 和 gID002 的块体构形文件分别为最初和最终的结构,弛豫轨迹文件 ID 为 gID001。
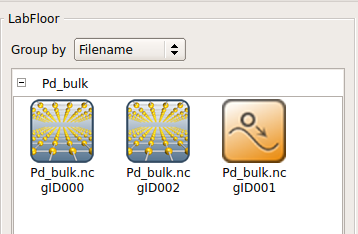
您可以拖拽这些文件到 Viewer ![]() 实现结构可视化。在浏览器中,将鼠标停在晶胞边界可以查看晶格常数,也可以在最新生成的 log 文件中查到。
实现结构可视化。在浏览器中,将鼠标停在晶胞边界可以查看晶格常数,也可以在最新生成的 log 文件中查到。
在接下来的计算中,您所用到的都是都是弛豫后的块体结构 gID002。
构造 Pd(100) 面并弛豫
- 拖拽弛豫后的块体构形到 Builder
 。
。 - 打开 Builders Surface (Cleave) 插件。
- 保留默认的 Miller 指数,单击 Next。
- 保留默认的 1×1 表面晶格,单击 Next。
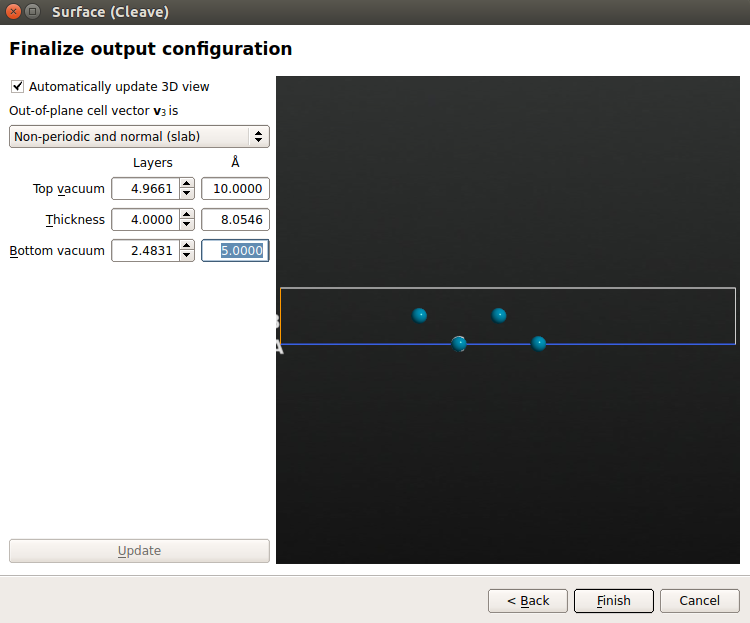
- 在 Out-of-plane cell vector 下方选择 Non-periodic and normal(slab),4 层平面金属,顶部和底端分别有 10 Å 和 5 Å 的真空。单击 Finish 创建表面。
- 发送板状结构到 Script Generator ,脚本中添加 New Claculator, OptimizeGeometry 和
 TotalEnergy 模块,修改默认输出文件名称为 Pd100.nc。
TotalEnergy 模块,修改默认输出文件名称为 Pd100.nc。
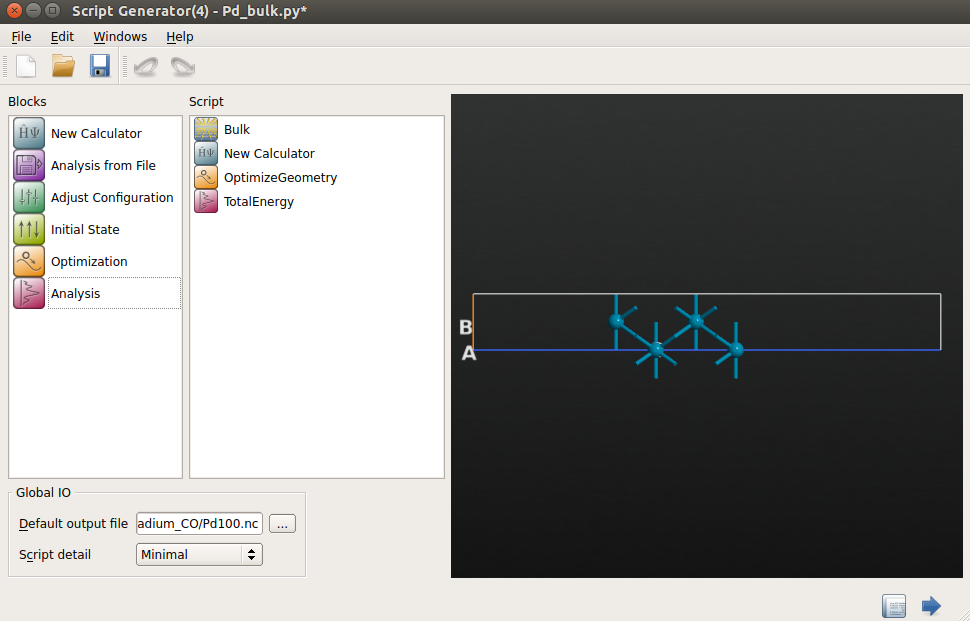
- 按照以下设置计算器参数:
- k-point sampling:9×9×1 k 点;
- exchange-correlation:GGA-PBE;
- Poisson solver 选择 FFT2D,平板之下设置 Neumann 边界条件,之上用 Dirichlet 边界条件。
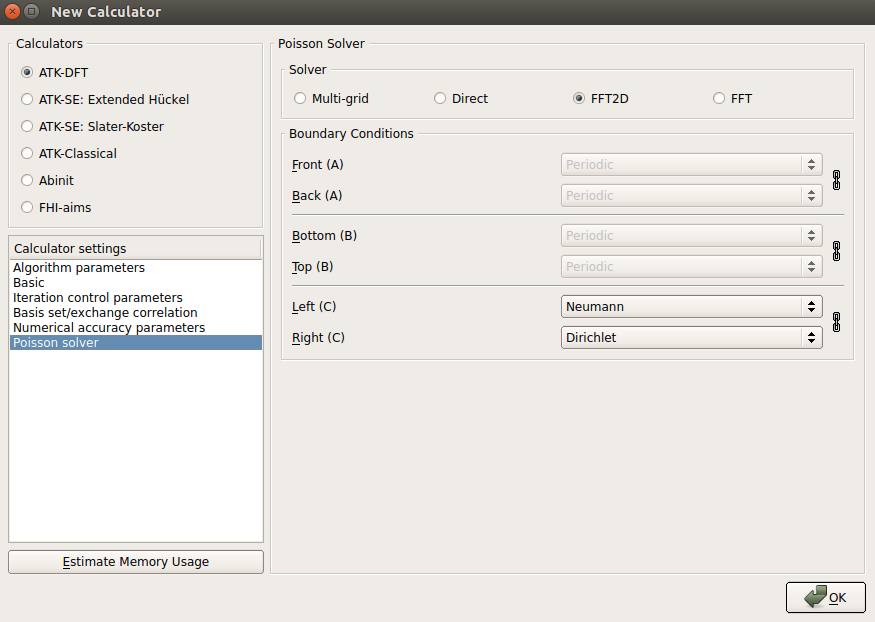
提示
这种沿模拟晶胞 C 方向上的特定边界条件设置很适合板状结构的计算,板上的真空在原则上可以扩展到无限远。
- 打开 OptimizeGeometry,为力的弛豫做出如下设置:
- 把轨迹保存到 Pd100_traj.nc 文件;
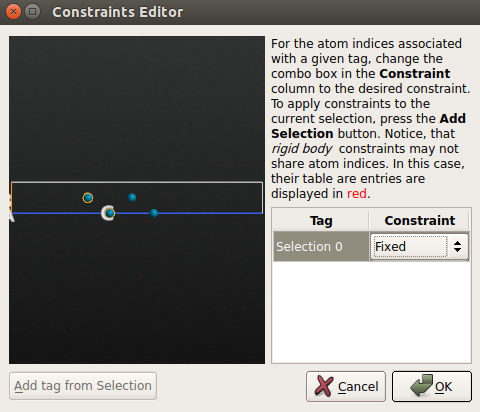
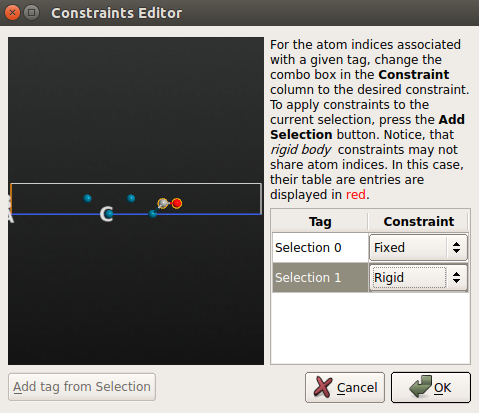
- 点击 Add Constraints 打开 Constraints Editor;
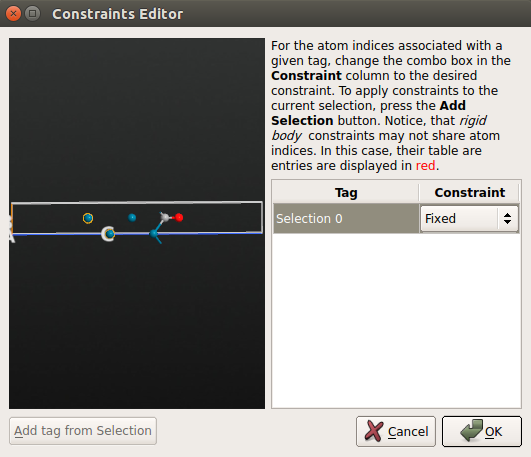
- 用鼠标选中 Pd(100) 面最底部的 2 层,点击 Add tag from selection 中。那两个原子现在被标注为 “Selection 0”,然后为其选择 Fixed 约束。
- 在 TotalEnergy 模块没有需要编辑的,所以到这步脚本已经完成了。保存为
Pd100.py,用 Job Manager 执行计算,大概需要 1 分钟。如果有需要,您可以在这 ↓Pd100.py 下载脚本。
执行计算,大概需要 1 分钟。如果有需要,您可以在这 ↓Pd100.py 下载脚本。
Pd(100) 面的结构优化完成后,结果会立即出现在 LabFloor。您还可以用 Viewer ![]() 把弛豫轨迹可视化。
把弛豫轨迹可视化。
提示
选中 TotalEnergy,点击 Text Representation 插件可以查看弛豫后 Pd(100) 面的总能量,这个能量值在 log 文件中也有输出。
弛豫 CO/Pd(100) 体系
下一步就是在表面上添加 CO 分子,然后弛豫整个体系。您可以下载现成的 CO/Pd(100) 结构 ↓CO_Pd100.py。您还可以通过教程 Building molecule-surface systems:Benzene on Au(111) 学习怎样在表面上添加分子。
正如在介绍中提到的,您需要两步来弛豫 CO/Pd(100) 构形:
- 为了快速了解 CO 被吸附的位置,给 CO 施加 Rigid 约束,面施加 Fixed 约束,并设置一个相对较低的电子密度的网格截断。
- 最后一步的弛豫不会约束 CO 分子,只固定 Pd(100) 板的底部。
精确弛豫
将提供的 CO/Pd(100) 构形(↓CO_Pd100.py)移动到 Script Generator,添加 ![]() New Claculator 和
New Claculator 和 ![]() OptimizeGeometry 模块。输入默认的输出文件名称为
OptimizeGeometry 模块。输入默认的输出文件名称为 Pd100_CO_rigid.nc,然后编辑脚本:
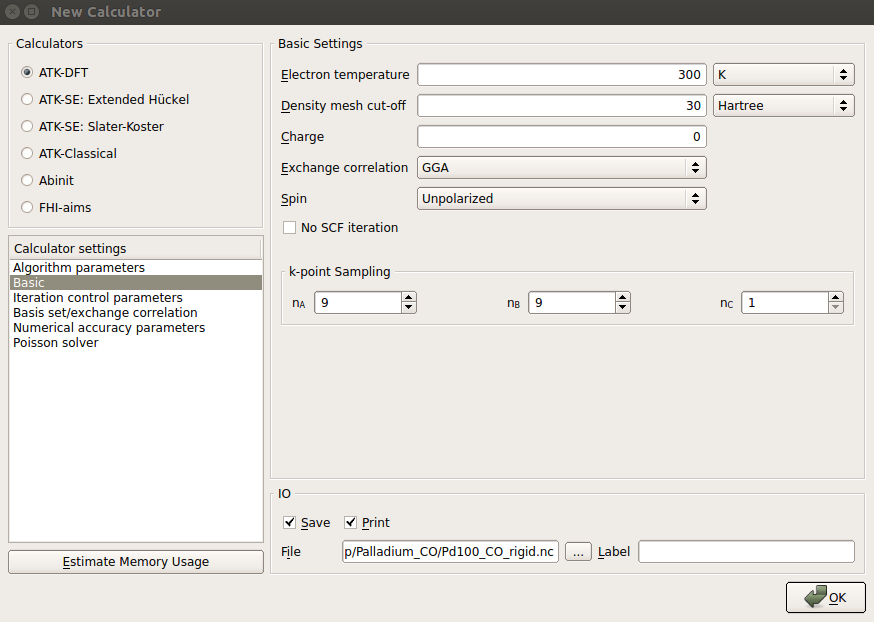
- 在 New Claculator 模块,除了将密度网格截断降至 30 Hartree 外,其他参数使用跟之前弛豫 Pd(100) 时相同的设置。
- OptimizeGeometry 模块的设置也与弛豫 Pd(100) 时相似,只是约束的选择有略微的不同:
- 给金属板的所有原子加上 Fixed 约束;
- CO 分子施加 Rigid 约束(把 C 原子和 O 原子添加到组 “Selection 1”)。
注意
在力优化时,被施加了 Rigid 约束的原子团在移动时像刚体。
- 保存脚本为
Pd100_CO_rigid.py,用 Job Manager 运行。如果串行执行的话大概耗时 5 分钟。您还可以在此 ↓Pd100_CO_rigid.py 下载脚本。
重要
如果您查看弛豫的轨迹会发现,CO 分子稍微偏移了初始位置,但 C-O 键的长度并没有改变,分子也未旋转。这就是刚性约束的作用。
最终弛豫
在此,您将以上一步得到的结果为初始状态,对 CO/Pd(100) 结构进行最终的结构优化。
- 把
Pd_CO_rigid.nc里 ID 为 gID002 的结构发送到 Scripter,默认输出文件名设为Pd_CO.nc。 - 再次添加 New Claculator, OptimizeGeometry 和 TotalEnergy 模块,参数编辑与之前步骤中的相似。只是,这次的 ATD-DFT 计算器的网格截断用 75 Hartree,且只对底部的 2 个 Pd 原子加 Fixed 约束。
- 保存脚本为
Pd_CO.py,在 Job Manager 或终端执行运算。模拟一共需要约 20 分钟,但如果在更多的 CPU 上并行运行的话,过程会快很多。脚本可以在此下载:↓Pd100_CO.py。
提示
在计算运行时,您可以用 Viewer ![]() 工具监控结构优化的进程。随着弛豫步数的增加,轨迹对象也在持续更新。
工具监控结构优化的进程。随着弛豫步数的增加,轨迹对象也在持续更新。
计算完成时,您应该看下完整的弛豫轨迹,可以观察到 CO 分子从在 Pd(100) 面上中空的吸附点附近移动到两个 Pd 表面原子间的桥位。

弛豫CO分子
为了计算得到 Pd(100) 面上 CO 的吸附能,您也需要弛豫单独的 CO 分子。我们可以通过以下步骤获得:
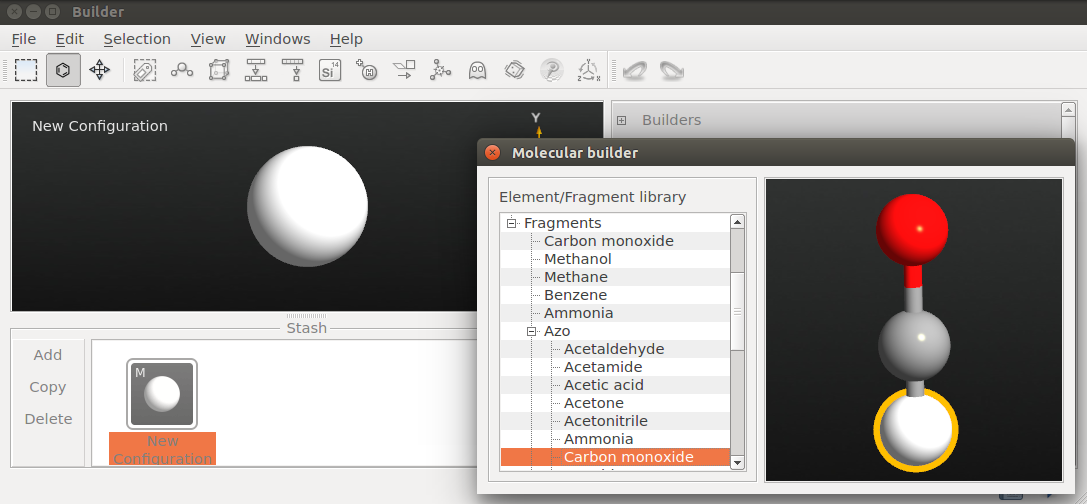
- 在 Builder 里点击 Add New Configuration 添加新的结构;
- 选择 Molecular Builder,在打开的面板中点击 Fragments Carbon monoxide;
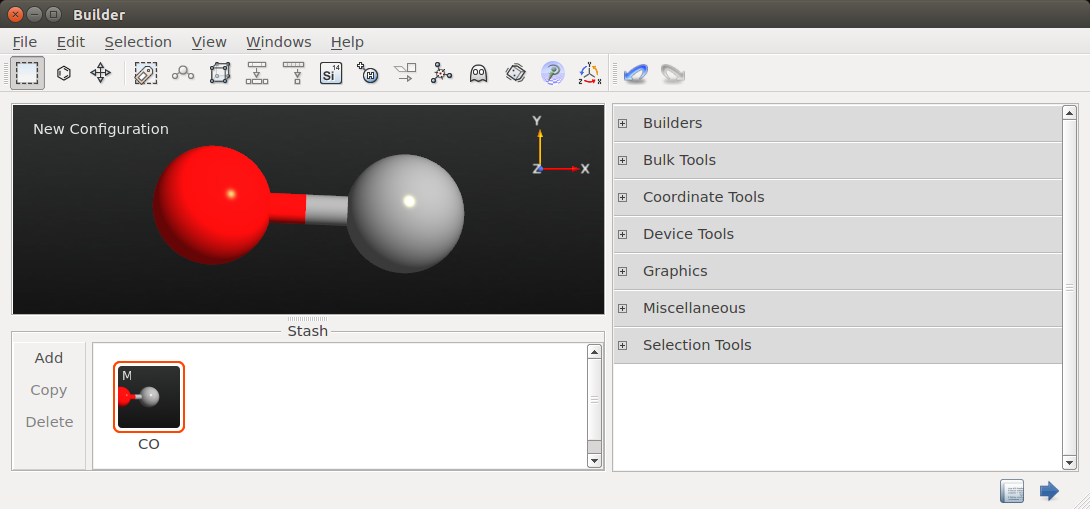
- 下一步,选择
New Configuration的 Stash 里的 H 原子,将其替换为 CO 片段,关闭 Molecular Builder 窗口,将 Stash 里文件名称更改为CO。
提示
想要了解更多关于怎样使用 Molecular Builder 的信息,您可以查看教程: Molecular Builder。
现在,您只需要弛豫 CO 原子坐标、计算总能量。把结构发送到 Script Generator,添加 ![]() New Claculator,
New Claculator,![]() OptimizeGeometry 和
OptimizeGeometry 和 ![]() TotalEnergy 模块。默认输出文件名称处输入 CO.nc,然后按照以下对模块稍作编辑:
TotalEnergy 模块。默认输出文件名称处输入 CO.nc,然后按照以下对模块稍作编辑:
- New Claculator:选择 GGA-PBE 泛函;
- OptimizeGeometry:您可以保存轨迹文件或减小 force tolerance,但这不是强制性的。
- 将脚本保存为 CO.py 并运行,计算速度非常快。脚本也可以在此处下载:↓CO.py。
您将会发现 C-O 键长的弛豫结果为 1.143 Å,与实验数据非常吻合。
吸附能
现在您已经准备好在完全的单分子层覆盖下计算 CO/Pd(100) 的吸附能,$\Delta E$ 由总能差得到:
$$\Delta E = E_\mathrm{products} - E_\mathrm{reactants} = E_\mathrm{Pd(100)/CO} - E_\mathrm{Pd(100)} - E_\mathrm{CO}$$
您可以用 Text Representations 插件查看这三个能量值,从而得到 $\Delta E$,或者还可以用脚本实现: ↓adsorption_energy.py。
计算吸附能时,采用 PBE 和含有 DZP 基组的 FHI 赝势,结果为 $\Delta E$ = -1.97 eV。但是,这个结果被所谓的基组叠加误差影响,所以您应该继续进行以下部分解决这个问题。
平衡校正
基组叠加误差(BSSE)是由 LCAO 基组的不完全造成的,会对不同子体系间的能量差产生重大影响。正如在教程 (BSSE counterpoise correction)中做出的解释,在 CO/Pd(100) 体系中 Pd 和 CO 基组的叠加会增加由人为因素造成的总能量降低,这是因为 Pd(100) 和 CO 子体系可以互相“借”基函数。结果就是吸附能太大。
中和 BSSE 的标准做法是应用所谓的平衡校正。在 ATK 里,通过使用 CounterpoiseCorrected 实现。
因此,您应该计算 CO/Pd(100) 体系的平衡(CP)校正总能量,以便计算得到的 CP 校正吸附能。
$$\Delta E^\mathrm{CP} = E^\mathrm{CP}_\mathrm{Pd(100)/CO} - E_\mathrm{Pd(100)} - E_\mathrm{CO}$$
采用如下所示的脚本(可在此下载:↓Pd_CO_cp.py)。该脚本读取之前弛豫的 CO/Pd(100) 结构和使用的计算器,并创建新的计算器应用在 CP 校正上。然后弛豫结构,计算总能量。
可以用 Job Manager 或终端运行脚本。它应该只执行 4 个非常小的弛豫步骤,在 10 分钟内完成计算。CP 校正会增大 CO/Pd(100) 体系的能量,导致吸附能 $\Delta E^\mathrm{CP}$ = -1.68 eV。
- Pd_CO_cp.py
1 # ------------------------------------------------------------- 2 # Bulk Configuration 3 # ------------------------------------------------------------- 4 5 bulk_configuration = nlread('Pd100_CO.nc', BulkConfiguration)[-1] 6 7 # Add tags 8 bulk_configuration.addTags('CO', [4, 5]) 9 bulk_configuration.addTags('Pd', [0, 1, 2, 3]) 10 bulk_configuration.addTags('Selection 0', [0, 1]) 11 12 # ------------------------------------------------------------- 13 # Calculator 14 # ------------------------------------------------------------- 15 16 # Get the calculator settings from non-BSSE calculation. 17 calculator = bulk_configuration.calculator() 18 basis_set = calculator.basisSet() 19 exchange_correlation = calculator.exchangeCorrelation() 20 numerical_accuracy_parameters = calculator.numericalAccuracyParameters() 21 poisson_solver = calculator.poissonSolver() 22 23 # Create BSSE calculator. 24 bsse_calculator = counterpoiseCorrected(LCAOCalculator, ["CO", "Pd"]) 25 calculator = bsse_calculator( 26 basis_set=basis_set, 27 exchange_correlation=exchange_correlation, 28 numerical_accuracy_parameters=numerical_accuracy_parameters, 29 poisson_solver=poisson_solver, 30 ) 31 32 bulk_configuration.setCalculator(calculator) 33 nlprint(bulk_configuration) 34 bulk_configuration.update() 35 nlsave('Pd100_CO_cp.nc', bulk_configuration) 36 37 # ------------------------------------------------------------- 38 # Optimize Geometry 39 # ------------------------------------------------------------- 40 constraints = [0, 1] 41 42 bulk_configuration = OptimizeGeometry( 43 bulk_configuration, 44 max_forces=0.05*eV/Ang, 45 max_steps=200, 46 max_step_length=0.2*Ang, 47 constraints=constraints, 48 trajectory_filename='Pd100_CO_cp.nc', 49 disable_stress=True, 50 optimizer_method=LBFGS(), 51 ) 52 nlsave('Pd100_CO_cp.nc', bulk_configuration) 53 nlprint(bulk_configuration) 54 55 # ------------------------------------------------------------- 56 # Total Energy 57 # ------------------------------------------------------------- 58 total_energy = TotalEnergy(bulk_configuration) 59 nlsave('Pd100_CO_cp.nc', total_energy) 60 nlprint(total_energy) ''