
这是本文档旧的修订版!
载流子迁移率
关键词:迁移率,电声耦合(电子-声子相互作用),温度相关载流子输运性质,电导率(conductivity),畸变势(deformation potential)
在本教程中吗,我们将介绍如何计算石墨烯(graphene)中的声子限制的电子迁移率。结合电子态和玻尔兹曼输运方程,同时考虑声子模式与电子-声子相互作用,使用 DFT 方法计算电子迁移率。我们将使用 QuantumATK 新版中提供的分析模块 ElectronPhononCoupling,DeformationPotential 和 Mobility 功能来完成。
计算迁移率 $\mu$ 的关键是计算电子的弛豫时间 $\tau$。弛豫时间有两种计算方法:
- 全角度(k,q)-相关:这种方法完整的考虑 $\tau$ 对电子和声子的波矢 $\mathbf{k}$ 和 $\mathbf{q}$ 的相关,即 $\tau=\tau(\mathbf{k},\mathbf{q})$。下面称为(k,q)-相关方法。
- 各向同性散射:这种方法仅考虑能量有关的 $\tau=\tau(E)$,假定 $\tau(E)$ 在动量空间中的变化是各向同性的。下面称为E-相关方法。
下面的计算表明,两种方法得到的结果相差无几,但是第二种方法比第一种方法计算速度大大提高。
文末将介绍理论背景知识和更多相关信息,已发表结果参见[GMSB16]。
提示
本教程使用特定版本的QuantumATK创建,因此涉及的截图和脚本参数可能与您实际使用的版本略有区别,请在学习时务必注意。
结构弛豫与电子态计算
在 Builder 中添加一个石墨烯结构:
- Add → Database → 搜素 “Graphene”
设置晶格常数C方向为20 Å:
- Bulk Tools → Lattice parameters, c=20Å
将结构在视图中居中(Coordinate Tools → Center → Apply),发送至脚本生成器Script Generator。
要进行精确的弛豫时间和迁移率计算,首先要精确的优化A、B方向的晶格常数并计算能带。
- 新增一个 New Calculator
- 设置 Opcupation methond 为 Methfessel-Paxton
- 设置 K-point sampling:kA=33,kB=33,kC=1
- 设置 Density mesh-cutoff 为 90 Ha

- 点击 Optimization,增加一个 OptimizeGeometry 模块,按如下设置参数:
- 最大受力 Force Tolerance 为 0.001eV/Å
- 取消勾选Constrain Lattice Vectors 后面的 x 和 y

- 添加 Analysis → Bandstructure
- 设置 Points per segment 为 100
- 设置 Brillouin Zone route 为 G, K, M, G
- 设置默认输出文件名 Default output file:
graphene_relax.hdf5 - 发送脚本到任务管理器 Job manager,运行计算任务
计算结束后,点击 LabFloor 中包含在 Graphene_relax.hdf5 的 Bandstructure 图标,使用右侧的 Bandstructure Analyzer 可以查看能带图。

将鼠标防止在一条能带上就可以看到相关的信息。比如上图中高亮的一条价带的编号为3,导带为4。这两条能带是与迁移率有关的关键能带,接下来我们将专注于这两条能带。
石墨烯中的声子
下面我们将计算石墨烯的动力学矩阵(dynamical matrix)。由动力学矩阵,可以得到声子谱和振动模式,从而可以测验计算的精确度。
步骤如下:
- 打开 LabFloor 中的
graphene_relax.hdf5文件,发送弛豫后的原子结构文件(gID001)到脚本生成器 Script generator,进行如下设置: - 设置 K 点网格:kA=3,kB=3,kC=1。
- 添加 Analysis → DynamicalMatrix 模块,设置参数如下:
- 选择 Repetitions 为 Custom
- 设置 nA=11,nB=11,nC=1

- 添加 Analysis → PhononBandstructure,设置如下参数:
- Points per segment:100
- Brillouin zone route:G,M,K,G
在 Sciprt Generator 主面板中,将 Default output file 改为:Graphene_dynmat.hdf5,脚本发送到 Job Manager,保存输入文件Graphene.py,完成计算。
为确保石墨烯电子态单胞计算时的k点采样与此处声子计算时的超胞一致,有必要设置 $\mathrm{k_i^{unit cell}} = \mathrm{k_i^{supercell}} \times \mathrm{N_i^{supercell}}$ ($\mathrm{i} = \mathrm{A},\mathrm{B},\mathrm{C}$)。
由于$11 \times 11$超胞尺寸较大(242原子),因此计算动力学矩阵非常耗时。不过,计算可以将原子位移并行化。由于单胞包含两个原子,每个原子在x,y,z三个方向上位移,因此有6个计算要完成。当总的计算数目乘以计算所用参数Proecesses per displacement等于计算所用的总核数时,计算效率最高。比如此例子中,如果设置Proecesses per displacement = 4,计算在24核心上完成只需要 30 分钟。
计算结束后,使用 Bandstructure Analyzer 查看声子谱结构,如下:
 图34. 声子谱结构。有 3 个声学模式:最下方一条,平面外模(ZA)在长波极限下∝q2,另外两条在小波矢附近与 q 线性相关。横声学模(TA)的能量低于纵声学模(LA)。
图34. 声子谱结构。有 3 个声学模式:最下方一条,平面外模(ZA)在长波极限下∝q2,另外两条在小波矢附近与 q 线性相关。横声学模(TA)的能量低于纵声学模(LA)。
石墨烯的迁移率
接下来,我们来看如何计算石墨烯中的电子迁移率。在已经计算的电子态结构和动力学矩阵的基础上,要计算迁移率还需要三步:
- 计算哈密顿量导数
- 计算电子-声子耦合
- 计算迁移率
其中第2、第3步分别有(k,q)-相关方法和E-相关方法两种,下文分别介绍。
哈密顿量微分
为了计算电子-声子耦合,需要首先计算哈密顿量的一阶偏微分。哈密顿量对第 $i$ 个原子的笛卡尔坐标$\alpha$的一阶偏微分,$\partial \hat{H}/ \partial R_{i,\alpha}$,可以使用 HamiltonianDerivatives 对象计算得到。
现在,按下面步骤操作:
- 打开 LabFloor 中的
graphene_relax.hdf5文件,发送弛豫后的原子结构文件(gID001)到脚本生成器 Script generator。 - 在 New Calculator,设置 K 点网格:(3,3,1)
- 添加 Analysis → HamiltonianDerivatives 模块,设置 nA=11,nB=11,nC=1.
- 设置默认输出文件名Default output file:
graphene_dHdR.hdf5
注意
此处的超胞重复次数需要和计算动力学矩阵的一致。
发送任务到任务管理器 Job manager,保存输入文件graphene_dHdR.py,运行计算任务。
注意
与动力学矩阵类似,哈密顿量导数计算相当耗时,但是可以将原子位移在多核上并行。此例子在24核上并行的计算时间大约1小时。
(k,q)-相关方法
要计算弛豫时间 $\tau(\mathbf{k},\mathbf{q})$ 和迁移率 $\mu$,需要在 $\mathbf{k}$- 和 $\mathbf{q}$-点的精细网格上计算电子-声子耦合。
电声耦合
单击Script Generator,添加如下计算单元:
- Analysis from File,打开选择文件
Graphene_relax.hdf5,加载其中Object Id为‘BulkConfiguration_1’的BulkConfiguration; - Analysis → ElectronPhononCoupling;
以下两个计算单元自动添加:
- Analysis → DynamicMatrix
- Analysis → HamiltonianDerivatives
在本例里,由于动力学矩阵和哈密顿量微分都已经计算过了,因此可以将这两个计算删去,添加两个Analysis from File,直接使用已有的结果。
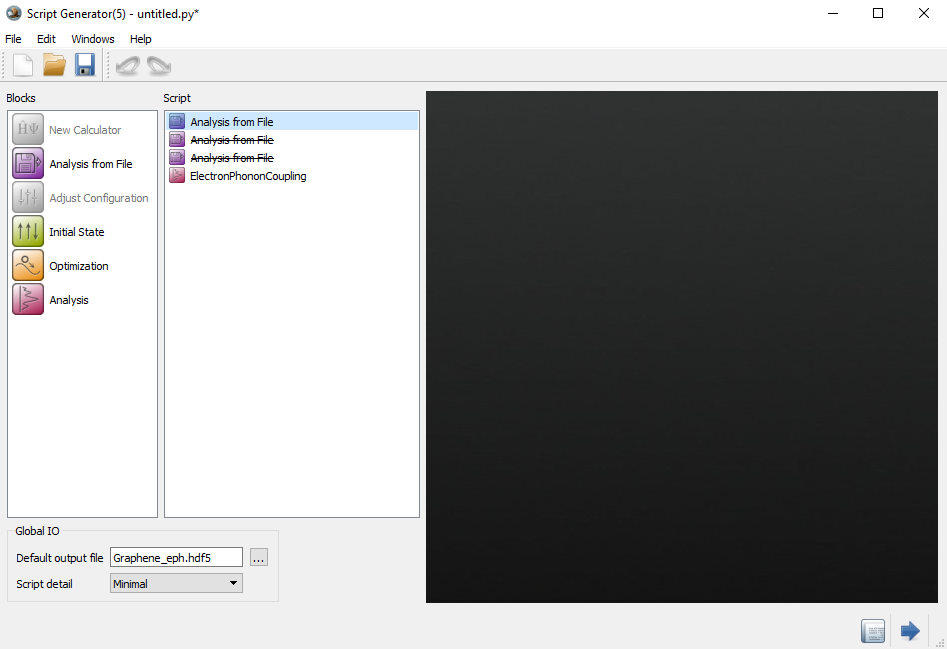
打开第二个Analysis from File,选择Graphene_dynmat.hdf5,加载DynamicMatrix数据对象,如下图:

打开第三个Analysis from File,选择Graphene_dHdR.hdf5,加载HamiltonianDerivatives数据对象,如下图:
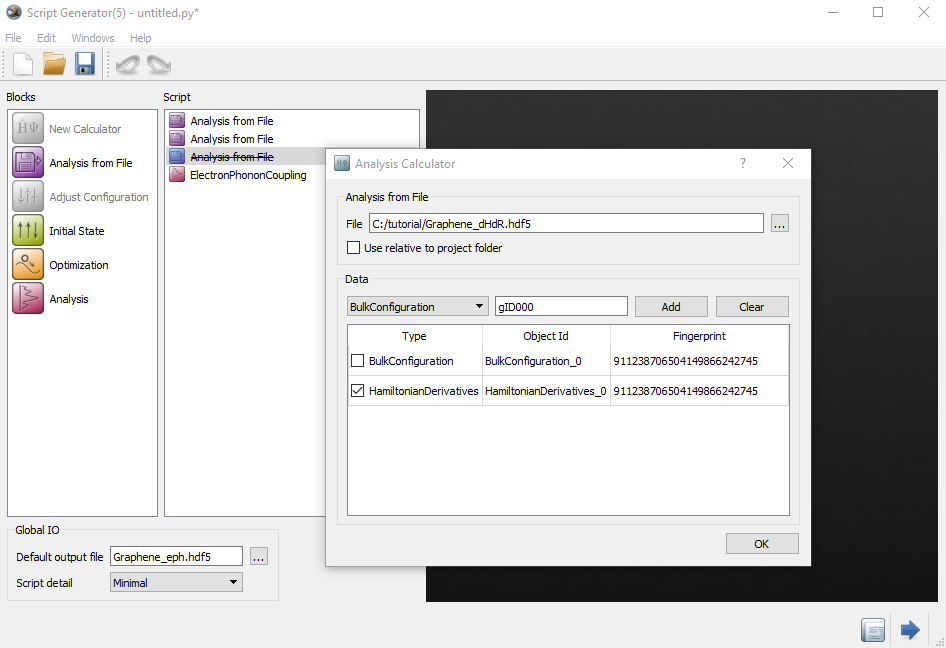
最后在ElectronPhononCoupling里设置如下参数:
- 在k-point sampling里,设置k grid为Custom(Cartesian),按下图设置min、max和number;
- 在q-point sampling里,设置k grid为Custom(Cartesian),按下图设置min、max和number;
- 选择Electron Band为3,4。
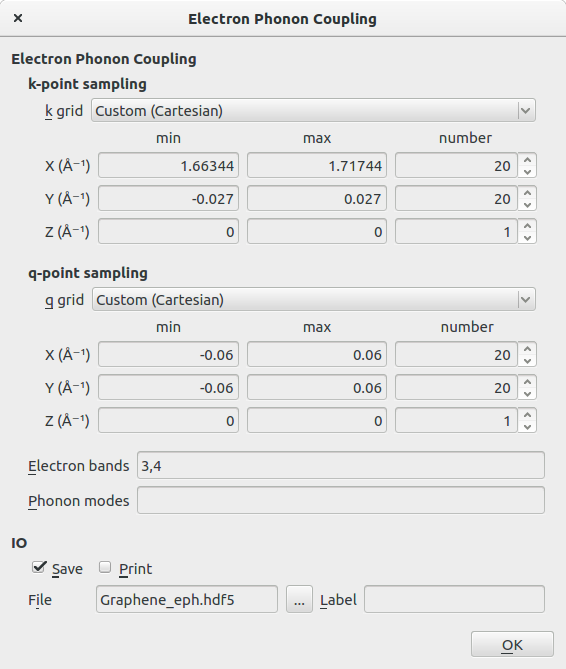
k和q的采样范围选择为仅在布里渊区的相关区域。对于前者是在$K$-点([1.7063,0.0,0.0])附近,后者是在$\Gamma$-点([0.0,0.0,0.0])附近。
在Script Generator主面板里设置输出文件为Graphene_eph.hdf5,将脚本发送至Editor,在下面一段按如下进行修改:
electron_phonon_coupling = ElectronPhononCoupling( configuration=bulk_configuration, dynamical_matrix=dynamical_matrix, hamiltonian_derivatives=hamiltonian_derivatives, kpoints_cartesian=kpoints, qpoints_cartesian=qpoints, electron_bands=[3,4], phonon_modes=All, energy_tolerance=0.01*eV, initial_state_energy_range=[-0.5,0.5]*eV, //修改这一行 store_dense_coupling_matrices=False, //修改这一行 )
对$k$- 和 $q$-空间进行高密度采样意味着计算非常耗时。合理设置energy_tolerance和initial_state_energy_range两个参数可以让计算仅考虑相关的电子-声子耦合矩阵元, 这样可以大大减少计算时间和对内存的需求。本例的计算在24核服务器上大约需要1.5小时。有关这两个参数的详细说明参见手册:ElectronPhononCoupling
将脚本发送到Job Manager,保存输入文件为Graphene_eph.py,完成计算。
迁移率
在LabFloor里,选择Graphene_relax.hdf5文件,将其中的Object Id ‘gID001’发送至Script Generator,添加如下计算单元,并修改参数:
- Analysis→Mobility
- Method 选择 Full angular (k,q)-dependence
- 选择 Fermi Shift 为 0.13 eV
- 不勾选 Calculate Hall coefficient
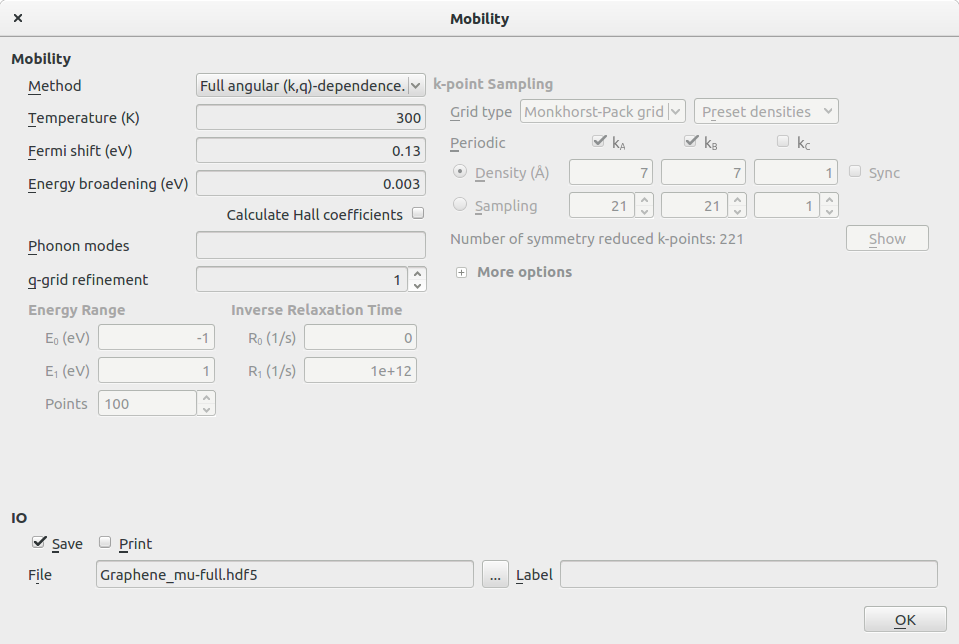
Fermi shift 0.13 eV相当于载流子浓度$n=10^{12}\mathrm{cm}^{-2}$
在Script Generator主面板里,将 Default output file 设为Graphene_mu-full.hdf5,将脚本发送至Job Manager,保存输入文件Graphene_mu-full.py,完成计算。
计算一旦完成,在LabFloor里Graphene_mu-full.hdf5,选择$\mu_{e,h}$图标,点击Text Representation。
+------------------------------------------------------------------------------+ | Mobility Report | | ---------------------------------------------------------------------------- | | Input parameters: | | Temperature = 300.00 K | | Fermi level shift = 0.13 eV | | Energy broadening = 0.0030 eV | | q-grid refinement = 1 | | | +------------------------------------------------------------------------------+ | Trace of linear responce tensors: | +------------------------------------------------------------------------------+ | | | Electrons: | | | | Mobility = 2.56e+05 cm^2/(V*s) | | Conductivity = 7.71e+06 S/m | | Seebeck coefficient = -6.11e-05 V/K | | Thermal conductivity = 4.46e+01 W/(m*K) | | Carrier density (2D, xy) = 3.76e+11 cm^-2 | | | | | | Holes: | | | | Mobility = 2.56e+06 cm^2/(V*s) | | Conductivity = 4.27e+04 S/m | | Seebeck coefficient = 1.81e-01 V/K | | Thermal conductivity = 4.02e+00 W/(m*K) | | Carrier density (2D, xy) = 2.09e+08 cm^-2 | | | +------------------------------------------------------------------------------+
计算得到的电子迁移率为$2.56 \cdot 10^{5}\ \mathrm{cm^{2} V^{-1} s^{-1}}$,与过去文献中在载流子浓度为$n = 10^{12} \mathrm{cm^{-2}}$和室温时的结果一致。
E-相关方法
电声耦合
下面我们用计算能量相关的弛豫时间的$\tau(E)$的方法来计算迁移率,为此我们需要重新计算电声耦合。我们假定弛豫时间在$\mathrm{k}$-空间是各向同性的。这意味着我们只需要从Dirac点(K点)向外计算一条线段上的电声耦合就足够了,很显然这样可以大大降低计算量。
单击Script Generator,添加如下计算单元:
- Analysis from File
- Analysis → ElectronPhononCoupling
与上一节一样从Graphene_dynmat.hdf读入动力学矩阵,从Graphene_dHdR.hdf5,读入哈密顿量微分。
在ElectronPhononCoupling单元,将参数作如下修改:
- 在 k-points sampling 里,设置k grid 为 Custom(Cartesian),采样的参数按如下设置。(注意到只有一条线段)
- 在 q-points sampling 里,设置q grid 为 Custom(Cartesian),采样的参数按如下设置。
- Electron Band 设为3,4
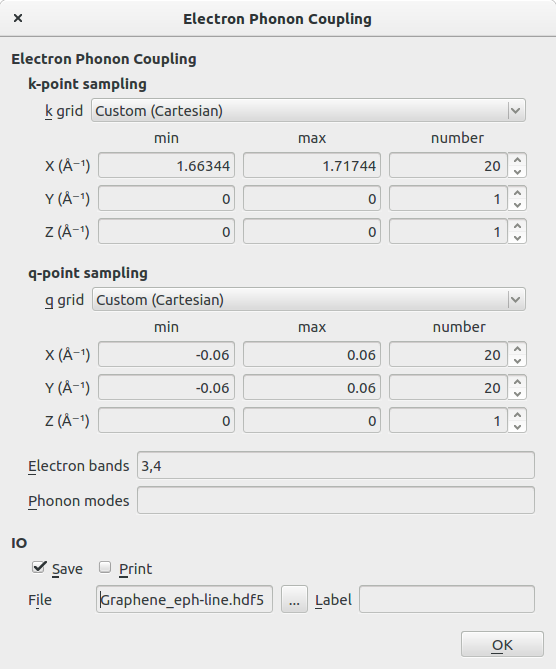
在Script Generator主面板里,将Default output file设置为Graphene_eph-line.hdf5,脚本发送至Editor,在ElectronPhononCoupling一段,作如下修改:
electron_phonon_coupling = ElectronPhononCoupling( configuration=bulk_configuration, dynamical_matrix=dynamical_matrix, hamiltonian_derivatives=hamiltonian_derivatives, kpoints_cartesian=kpoints, qpoints_cartesian=qpoints, electron_bands=[3,4], phonon_modes=All, energy_tolerance=0.01*eV, //修改这一行 initial_state_energy_range=[-0.5,0.5]*eV, //修改这一行 store_dense_coupling_matrices=False, )
脚本发送至Job Manager,保存输入文件为Graphene_eph-line.py,完成计算。
由于计算只对k空间的一个线段采样,计算比(k,q)-全相关方法大大提速,这个计算在24核服务器上计算大约只需要5分钟。
迁移率
现在我们可以用两步计算来得到基于能量相关的弛豫时间$\tau(E)$的室温下的迁移率$\mu$。
- 第一步,计算从K-点向外、沿着线段的$\mathrm{k}$- 和$\mathrm{q}$-相关的弛豫时间$\tau(\mathrm{k},\mathrm{q})$;
- 第二步,将$\tau(\mathrm{k},\mathrm{q})$对$\mathrm{k}$-空间进行平均,得到$\tau(E)$,用于计算迁移率$\mu$。
形变势计算
现在,我们已得到计算电子-声子耦合性质的要素:1)动力学矩阵;2)哈密顿量微分。首先,我们将在倒空间沿特定方向拟合形变势。然后,在 q 网格中计算电子-声子耦合,及声学声子极限下的迁移率。
首先,为得到声学模的形变势,我们考虑电子-声子耦合矩阵元随q的变化关系,$|M_{\mathbf{k},\mathbf{q}}^\lambda|$,操作如下:
- 打开 LabFloor 中的 graphene_relax.nc 文件,发送弛豫后的原子结构文件(gID001)到脚本生成器 Script generator。
- 添加分析器Analysis → DeformationPotential,
当添加 DeformationPotential 模块,DynamicalMatrix 和 HamiltonianDerivatives 将被自动添加到脚本生成器中,得到如下:

查看计算器 Calculator 中 k-point 取 (7,7,1),而不是计算动力学矩阵和哈密顿量微分时使用的 k 点 (1,1,1)。
现在,打开 DeformationPotential,首先设置电子初态的 k-point,设置如下:
- 初始设置k-point = K。在K点(狄拉克点,Dirac point)右侧,导带和价带简并
- 设置 k-point = Other(Fractional)
- 设置 a = b = 0.33334
K点右侧价带(valence band, VB)和导带(conduction band, CB)简并。这表明,任何两个态的线性组合都是电子本征态。由于价带和导带有不同的对称性,他们耦合为不同的声子。波矢稍稍偏离 K 点时,我们假设为纯价带和纯导带。
下面,需要指定考虑的声子 q 点值。大多情况下,我们对决定形变势和迁移率的小$|q|$值感兴趣。我们可以通过由一个初始 q 点和方向矢量来确定一条线上的 q 点值。初始 q 点和方向矢量由分数坐标给出。我们可以选取布里渊区的一个路径。使用前面的参数:
- 保持 First point 为 $\Gamma$ 点
- 设置方向矢量:a, b, c = (0.1, 0, 0)
- 设置每段的 q 点数:20
- 设置电子能带:3,4
 再增加一个 DeformationPotential 对象,其他参数一样,除
再增加一个 DeformationPotential 对象,其他参数一样,除
- 方向矢量:a, b, c = (0.1, 0.1, 0)

- 设置默认输出文件名 Default output file: graphene.nc
由于我们已计算了动力学矩阵和哈密顿量微分,不必再重新计算,直接从文件读取数据即可。发送脚本到编辑器 Editor,并替换 Dynamical Matrix 和 Hamiltonian Derivatives 部分如下:
# ------------------------------------------------------------- # Dynamical matrix # ------------------------------------------------------------- dynamical_matrix = nlread('graphene.nc', DynamicalMatrix)[0] # ------------------------------------------------------------- # Hamiltonian derivatives # ------------------------------------------------------------- hamiltonian_derivatives = nlread('graphene.nc', HamiltonianDerivatives)[0]
发送计算任务至任务管理器 Job manager 并运行计算任务。任务耗时几分钟完成。
形变势分析
计算完成后使用 Deformation Potential Analyzer 分析器可视化 LabFloor 中的计算结果文件 graphene.nc。两张图分别显示了所选模式的声子能量随 $|q|$(上图)和电子-声子耦合矩阵元随 $|q|$(下图)的变化规律。图的下部显示有一些分析选项。可以选择如下:
- 初始和最终电子能带的指标
- 声子模式
- 耦合矩阵类型,unscaled 或 scaled
- 线性拟合区间
在 Fitting output 下方显示线性拟合结果,零阶、一阶形变势,以及线性拟合的质量。

试着改变不同的电子能带、声子模式、拟合区间,看看有什么变化。对比各 DeformationPotential 对象,q 取沿 (0, 0, 0)→(0.1, 0.1, 0)方向。分析计算数据,得到如下结论:
- 平面外声子模式(0)与电子价带(3)、导带(4)均不耦合。
- 在小 q 值处,声子模式 1 和 2(LA 和 TA)呈线性规律,表示一阶形变势模型的准确性。但是,拟合形变势值取决于特定的 q 方向。简化形变势近似中,假设球对称性,并由 q 决定,因此,应使用合适的平均形变势值。
- 尽管应考虑更多的方向来得到精确的期望值,TA 和 LA 声子模式的一阶形变势值仍在 2-5eV 区间内。
- LO 和 TO 光学模式(4,5)接近常数,用零阶形变势模型描述基本正确。
注意 有些情况下,可能会有“有趣”的声子能量或耦合矩阵跳跃。这通常是由于声子能带排序造成的。我们按照能量对声子模式进行排序。例如,LO 和 TO模式多次交叉,有时 LO 模式的指标是 4,有时该模式的指标是 5。这些跳跃和交叉可以通过绘制“所有”声子模式得到印证。
为更精确研究形变势随角度的变化,尝试下面的步骤:
- 退回到前面计算形变势的 python 脚本
- 删除最后的 DeformationPotential 模块
- 修改脚本中的 Deformation 部分的程序行,将下面的程序语句:
# ------------------------------------------------------------- # Deformation potential # ------------------------------------------------------------- q_0 = numpy.array([0, 0, 0]) q_direction = numpy.array([0.1, 0, 0]) points_per_segment = 20 q_path = [list(q_0 + s*q_direction) for s in numpy.linspace(0, 1, points_per_segment)]
替换为:
# ------------------------------------------------------------- # Deformation potential # ------------------------------------------------------------- # Load function to convert from cartesian to fractional coordinates. from NL.CommonConcepts.Configurations.Utilities import cartesian2fractional # Radius of circle, in units of Ang^-1. q_norm = 0.01 thetas = numpy.linspace(0,numpy.pi*2,60) # Setup list of cartesian q-points in a circle. q_circle_cartesian = [[q_norm*numpy.cos(t), q_norm*numpy.sin(t), 0] for t in thetas]*Ang**-1 # Get the reciprocal lattice vectors. reciprocal_vectors = bulk_configuration.bravaisLattice().reciprocalVectors() # Convert q-point to fractional. q_path = cartesian2fractional(q_circle_cartesian, reciprocal_vectors)
首先设置笛卡尔坐标系下圆形的一系列 q 点,然后转化为 q 点的分数坐标。运行脚本,查看 DeformationPotential 对象,如下图所示为初始能带和最终能带均为 4,声子模指标为 2(LA 模)的情况。电子-声子耦合矩阵元显示出明显的震荡。平均值接近 0.04 eV/Å。由于 q 圆的半径为0.01 $Å^{-1}$,一阶形变势平均值 $D^{(1)}$ 为 4eV,与文献 [KTJ12b] 报道一致。

电子-声子耦合
DeformationPotential对象中包含了计算迁移率所需的一些信息,即电子-声子耦合项。但是,为计算弛豫时间和迁移率,我们需要知道初始 k 值构成的有限 q 网格下的耦合项。这是由 ATK 中 ElectronPhononCoupling 分析对象处理的。
下面将使用两个 ElectronPhononCoupling 对象。首先,计算 k 空间中完整 q 网格下沿一条线上的耦合元,用来分析 ($q_x$, $q_y$) 点的耦合矩阵元,及特定能量和温度下的弛豫时间的计算。操作如下:
- 打开 LabFloor 中的
graphene_relax.nc文件,发送弛豫后的原子结构文件(gID001)到脚本生成器 Script generator。 - 添加分析器 Analysis → ElectronPhononCoupling
注意
与添加DeformationPotential对象一样,添加ElectronPhononCoupling对象时,DynamicalMatrix和HamiltonianDerivatives将被自动添加。
我们要计算 Δ 连线(由原点至 K 点)上 k 点的电子-声子耦合。笛卡尔坐标系中,K 点坐标为(1.69044, 0, 0),可以由电子能带结构Bandstructure 对象中得到该信息,即在 LabFloor 中点击 Text Representation。现在,打开 ElectronPhononCoupling 对象,设置如下:
- 自定义 k 网格,k grid = Custom(Cartesian)
- 自定义 q 网格,q grid = Custom(Cartesian)
- 电子能带,Electron bands = 3, 4
- 声子模式,Phonon modes = 1, 2(我们仅关注声学模)

运行计算前,发送脚本至编辑器 Editor,用下面的程序语句替换动力学矩阵和哈密顿量微分部分的语句:
# ------------------------------------------------------------- # Dynamical matrix # ------------------------------------------------------------- dynamical_matrix = nlread('graphene.nc', DynamicalMatrix)[0] # ------------------------------------------------------------- # Hamiltonian derivatives # ------------------------------------------------------------- hamiltonian_derivatives = nlread('graphene.nc', HamiltonianDerivatives)[0]
运行计算前,注意:
注意
我们将计算非常多(q,k)点上的电子-声子耦合,每个计算不需要很多内存,因此这个任务非常适合并行运行。128MPI 核运行需约 10 分钟,但若单核运算将耗费很长时间。
当计算结束后,使用Electron-Phonon Coupling Analyzer分析器可视化结果。显示如下图的窗口:

电子-声子耦合分析工具提供了丰富的信息,在工具窗口左侧你可以设置一些输入的参数用于作图:
- Electron-Phonon Coupling Analyzer 分析器包含很多信息。窗口左侧设置绘制图的输入参数:
- Initial k-point 和 Initial band 指标。“Initial State Properties” 栏下显示了对应 k 点和能量。
- Final band,决定您是否考虑了能带内或能带间散射。
- Phonon mode,声子模式指标。
- Coupling matrix,可设置为 “scaled”或“unscaled”, 与 Deformation Potential 中的定义一样,参见上文。
- Energy broadening,此参数决定了绘图窗口中$\delta$-函数的展宽。$\delta$-函数在程序中用 Lorentzian 函数 $\delta(E)\approx \frac{1}{2\pi}\frac{\Gamma}{\Gamma^2 + E^2}$。能量展宽决定了 $\Gamma$ 的值。取较小的能量展宽,将会得到较窄的$\delta$-函数。q 网格优化的设置限制了能够取多小的值。
- 最后,通过 Refinement 参数设置优化 q 点网格。因此,如果最初进行了 (20, 20, 1) q 网格的计算,通过设置 Refinement = 2 可以得到 (40, 40, 1) q 网格。所有与 q 有关的性质,在新的 q 网格中可以线性插值得到。增加 Refinement 可以使您选择更小的能量展宽值。
注意
优化参数和能量展宽是计算迁移率的输入参数。
Electron-Phonon Coupling Analyzer 中作图的所有物理量均以 $q_x$ 和 $q_y$ 为自变量。上面一行画出声子能量(左图),吸收声子的终态电子能量 $E_{k+q}$(中图),产生声子的终态电子能量 $E_{k-q}$(中图)。下面一行画出电子-声子耦合矩阵元(左图),吸收(中图)和发射(右图)声子的能量守恒 $\delta$-函数。
尝试调整绘图参数查看作图。
在开始迁移率计算之前,我们需要查看 Electron-Phonon Coupling Analyzer 中的:
- q-区间选取是否足够大?$\delta$-函数图中的环应被完全覆盖,而不是仅显示部分环。否则将不能覆盖散射相关的q区间。需要注意的是,通过改变初态的k指标,我们可以改变$\delta$-函数图中环的大小和形状。这是因为石墨烯布里渊区狄拉克点(Dirac cones)相应的发生移动。
- q 网格采样是否足够完备?鉴于计算迁移率时使用的温度(见下图),能量展宽通常应满足 $\Gamma < k_BT$。低温下,需要设置小的能量展宽和完备的 q 采样。无需重新计算电子-声子耦合,这个工作可以通过增加优化系数得到。
弛豫时间倒数
在计算实际迁移率之前,我们需要使用 Mobility 对象分析导带的弛豫时间 $\tau_{k,n}$随 $k$ 的变化关系。这在像石墨烯这样的二维材料中具有特别有趣的性质。根据 Bloch-Gruneisen 定理,由于相空间中的反向散射的减弱,弛豫时间在费米能级处显著增加。
- 打开 LabFloor 中的 graphene_relax.nc 文件,发送弛豫后的原子结构文件(gID001)到脚本生成器 Script generator。
- 添加分析器 Analysis → Mobility
打开 Mobility 对象,按照下图设置参数:

发送脚本到编辑器 Editor 中,删除上面 Mobility 部分的全部语句。这是由于使用已计算完成的数据,这部分就不需要了。添加下面的语句。
bulk_configuration = nlread('graphene.nc', BulkConfiguration)[1] electron_phonon_coupling = nlread('graphene.nc', ElectronPhononCoupling)[0] # ------------------------------------------------------------- # Mobility # ------------------------------------------------------------- mobility = Mobility( configuration=bulk_configuration, electron_phonon_coupling=electron_phonon_coupling, temperature=10*Kelvin, phonon_modes=[1], fermi_shift=0.15*eV, energy_broadening=0.003*eV, refinement=15, ) nlsave('graphene_mobility.nc', mobility)
计算大约耗时 10-20 分钟。如此长的计算时间,是由于为精确描述低温下的情况,需要密的优化网格。迁移率计算完成后,使用下面的脚本可视化弛豫时间倒数随能量的变化。
import pylab as pl mobility = nlread('graphene_mobility.nc', Mobility)[0] # The phonon mode that should be considered. mode = 1 # Get the inverse life time in ns^-1 gamma = mobility.inverseLifeTime().inUnitsOf(Second**-1)*1e-9 # Get the Fermi shift fermi_shift = mobility.fermiShift() # Get the eigenvalues at k. e_k = mobility.eigenvaluesK().inUnitsOf(eV) # Get Fermi level Ef = mobility._fermiLevel().inUnitsOf(eV) # Spin index i_spin = 0 # Bloch state index. The electron-phonon coupling was calculated for bands 3 and 4. We look at 4, which has index 1. i_bloch = 1 pl.figure(1) pl.plot((e_k[i_spin, :, i_bloch] - Ef)/fermi_shift.inUnitsOf(eV), gamma[i_spin, mode, :, i_bloch], 'o-') pl.xlabel('$\epsilon/\epsilon_F$', fontsize=14) pl.ylabel('Inverse Life Time (ns$^{-1})$', fontsize=14) pl.legend(loc=0) pl.xlim([0, 1.7]) pl.show()

图中弛豫时间倒数的下降(低散射率)对应费米能级处电子态满足 Bloch-Gruneisen 定理。二维系统中,低温下反向散射在相空间中被抑制,散射几率降低。这一效应的理论研究以前在其他二维电子气异质结[KS92]和类石墨烯材料[GMSB16], [HS08], [KTJ12], [KTJ12b]中也曾报道过。
迁移率
为了计算迁移率,需要在石墨烯狄拉克点 $K$ 附近进行 k 点采样,因此,需要建立一个新的 ElectronPhononCoupling 对象。操作如下:
- 打开 LabFloor 中的 graphene_relax.nc 文件,发送弛豫后的原子结构文件(gID001)到脚本生成器 Script generator。
- 添加分析器 Analysis → ElectronPhononCoupling
注意
与前面一样,添加ElectronPhononCoupling对象时,DynamicalMatrix和HamiltonianDerivatives对象将被自动添加进来。
打开ElectronPhononCoupling对象,设置如下图的参数:

运行计算前,将脚本发送至编辑器 Editor,用下面的语句替换 Dynamical Matrix 和 Hamiltonian Derivatives 段:
# ------------------------------------------------------------- # Dynamical matrix # ------------------------------------------------------------- dynamical_matrix = nlread('graphene.nc', DynamicalMatrix)[0] # ------------------------------------------------------------- # Hamiltonian derivatives # ------------------------------------------------------------- hamiltonian_derivatives = nlread('graphene.nc', HamiltonianDerivatives)[0]
8 MPI 进程并行计算大约需要 2 小时完成计算任务。
注意
正方形网格k和q点采样时,在比较大k空间区域考虑了非常多的k点,因此计算会非常耗时。取$20 \times 20$ k点和 $20 \times 20 $ q 点(即 $20^4=1.6 \times 10^5$ 个k或q点)时 8 核 MPI 并行运算大约耗时 2 小时。
增加 k 和 q 点采样为 $40 \times 40$ 网格($2.56 \times 10^6$ 个 k 或 q 点)时 8 MPI 并行运算大约耗时 1 天半。k 点的选取窗口对应能量的窗口在费米能级附近的 -0.15eV ~ 0.15eV 间,参见前面能带计算结果。
如果要计算能量窗口以外的迁移率,需要增加额外的 k 点,也耗费更多的计算时间。
此外,要计算低能量电子的迁移率,需要缩小k点的选取窗口,以便节省计算时间和确保足够高的网格密度。
计算完成后,按照下面步骤操作:
- 打开 LabFloor 中的 graphene_relax.nc 文件,发送弛豫后的原子结构文件(gID001)到脚本生成器 Script generator。
- 添加分析器 Analysis → Mobility
注意
添加 Mobility 对象时,DynamicalMatrix,HamiltonianDerivatives 和 EelctronPhononCoupling 对象将被自动添加进来。
打开 Mobility 对象,按照下图设置参数:

注意
载流子浓度 $n \approx 10^{12} cm^{-2}$ 时费米移动 0.13eV。
发送脚本至编辑器,删除上面的 Mobility 语句,并添加下面脚本中前两行(剩下的部分是迁移率对象,与脚本中一致)。
bulk_configuration = nlread('graphene.nc', BulkConfiguration)[1] electron_phonon_coupling = nlread('graphene.nc', ElectronPhononCoupling)[1] # ------------------------------------------------------------- # Mobility # ------------------------------------------------------------- mobility = Mobility( configuration=bulk_configuration, electron_phonon_coupling=electron_phonon_coupling, temperature=300*Kelvin, phonon_modes=All, fermi_shift=0.13*eV, energy_broadening=0.003*eV, refinement=1, ) nlsave('graphene.nc', mobility)
计算约耗时几秒钟。
计算完成后,打开 LabFloor 中的 graphene.nc 文件,找到迁移率对象,点击 Text Information 按钮。计算得到的电子迁移率$\approx 2 \times 10^5 cm^2/Vs$ 与文献报道的室温下电子迁移率 $n \approx 1 \times 10^{12} cm^{-2}$ 结果一致[KTJ12b]。
为得到石墨烯形变势的值,可以由 DFT 方法计算迁移率随温度的变化,然后通过($\ref{7}$)式拟合得到。迁移率计算完成后,迁移率随温度变化关系的简单计算脚本如下:
- plot_mu_t.py
# Load packages from pylab import * from scipy.optimize import curve_fit # Read in the relaxed structure and the electron-phonon coupling bulk_configuration = nlread('graphene_relax.nc', BulkConfiguration)[-1] electron_phonon_coupling = nlread('graphene.nc', ElectronPhononCoupling)[-1] # Define parameters for graphene fermi_velocity = 1e6*Meter*Second**(-1) sound_velocity = 14.1*1e3*Meter*Second**(-1) mass_density = 7.6*1e-7*kiloGram*Meter**(-2) fermi_shift = 130*meV carrier_concentration = (130.0/11.65)**2*1e14*Meter**(-2) # Analytical mobility as a function of temperature, see Eq. (7) def f(temperature, D): """ temperature: The temperature of the system with units of Kelvin. D : The deformation potential. No unit, but values in eV. """ mobility = 4*elementary_charge*hbar*mass_density*fermi_velocity**2*sound_velocity**2 / \ (numpy.pi*carrier_concentration*boltzmann_constant*(temperature*Kelvin)*(D*eV)**2) # Return mobility in units of cm^2/Vs return mobility.inUnitsOf(Meter**2/(Volt*Second))*1e4 # Loop over temperatures and calculate the mobility Ts = numpy.linspace(100.0, 300.0, 10) mu_list = [] for T in Ts: temp=T*Kelvin # ------------------------------------------------------------- # Mobility # ------------------------------------------------------------- mobility = Mobility( configuration=bulk_configuration, electron_phonon_coupling=electron_phonon_coupling, temperature=temp, phonon_modes=[1], fermi_shift=fermi_shift, energy_broadening=0.003*eV, refinement=1, ) mu_h,mu_e=mobility.evaluate() mu_list.append(mu_e.inUnitsOf(Meter**2/(Volt*Second))*1e4) # Plot the data plot(Ts,mu_list,'-o' ) # Fit the data D,tmp = curve_fit(f,Ts, mu_list) # Plot the fit plot(Ts,f(Ts,D[0]),label='Deformation Potential = %.2f eV'%D[0]) legend(loc=0) ylabel('Mobility [cm^2/(V*s)]') xlabel('Temperature [K]') show()
注意
由于解析式中仅考虑了单个有效声子模式,因此此拟合中我们仅考虑了 TA 石墨烯模式的贡献。参数来自文献中的解析模型[KTJ12b]。

拟合结果中明显可以看到迁移率与温度的反比 (~1/T) 的关系,与($\ref{7}$)式符合很好。计算得到的形变势 8.8eV 比已报道的结果略小[KTJ12b]。
理论知识
迁移率 $\mu$ 与电导率 $\sigma$ 有如下关系: $$ \mu = \frac{\sigma}{ne} \tag{1}\label{1}$$
$n$ 表示载流子浓度,$e$ 表示电子电荷。
本文中,我们将采用半经典玻尔兹曼输运方程(BTE)计算电导率。在零磁场和恒定电场下,定态玻尔兹曼方程简化为: $$\frac{q\mathbf{E}}{\hbar} \cdot \nabla_{\mathbf{k}} f_{\mathbf{k} n} \approx \frac{q\mathbf{E}}{\hbar} \cdot \nabla_{\mathbf{k}} f^0_{\mathbf{k} n} = \left.\frac{\partial f_{\mathbf{k}n}}{\partial t}\right|_{coll} \tag{2}\label{2}$$
方程($\ref{2}$)右侧,常被称为碰撞积分(collision integral),用来描述不同的散射机制。由多种机制散射后,系统可耗散至稳态。方程左侧与电场强度线性相关,比例系数由分布函数决定。
电声子散射由碰撞积分描述,通常采用弛豫时间近似: $$\left.\frac{\partial f_{\mathbf{k}n}}{\partial t}\right|_{coll} = \frac{\delta f_{\mathbf{k}n}}{\tau_{\mathbf{k}n}}$$
描述声学声子的准弹性散射(弛豫时间近似)。用 $\lambda$ 标记声子模式,$n$ 标记电子能带,$k$ 标记电子波矢。由 $|\mathbf{k}n\rangle$ 态到 $|\mathbf{k}'n'\rangle$ 态的跃迁几率可以由 费米黄金规则(FGR)得到:
$$\begin{split}P_{\mathbf{k}\mathbf{k'}}^{\lambda nn'} &= \frac{2\pi}{\hbar} |g_{\mathbf{k}\mathbf{k'}}^{\lambda n n'}|^2 [ n_{\mathbf{q}}^{\lambda} \delta \left(\epsilon_{\mathbf{k}'n'}-\epsilon_{\mathbf{k}n}-\hbar \omega_{q \lambda} \right) \delta_{\mathbf{k}',\mathbf{k}+\mathbf{q}} \\[.5cm] &+ (n_{\mathbf{-q}}^{\lambda}+1) \delta \left( \epsilon_{\mathbf{k}'n'}-\epsilon_{\mathbf{k} n}+\hbar \omega_{-q \lambda} \right) \delta_{\mathbf{k}',\mathbf{k}-\mathbf{q}} ],\end{split}$$
方括号中第一项和第二项分别表示吸收一个声子和放出一个声子。在 QuantumATK 中,使用 ElectronPhononCoupling 分析模块计算电声子耦合矩阵元 $ |g_{\mathbf{k}\mathbf{k'}}^{\lambda n n'}|$。从而得到电声子碰撞积分:
$$\begin{split}\left.\frac{\partial f_{\mathbf{k}n}}{\partial t}\right|_{coll} &= - \sum_{\mathbf{k}'n'} \left[f_{\mathbf{k}n}\left(1-f_{\mathbf{k}'n'}\right)P_{\mathbf{k}\mathbf{k}'}^{nn'}-f_{\mathbf{k}'n'}\left(1-f_{\mathbf{k}n}\right)P_{\mathbf{k}'\mathbf{k}}^{n'n}\right]\end{split} \tag{3}\label{3}$$
弛豫时间近似
弛豫时间近似,是采用非常简单的形式来表示碰撞积分: $$\begin{split}\left.\frac{\partial f_{\mathbf{k}n}}{\partial t}\right|_{coll} &=& -\frac{\delta f_{\mathbf{k}n}}{\tau_{\mathbf{k}n}}\end{split} \tag{4}\label{4}$$
其中 $\delta f_{\mathbf{k}n} = f_{\mathbf{k}n}-f^0_{\mathbf{k}n}$。因此,线性玻尔兹曼输运方程可写为: $$q \mathbf{E} \cdot \mathbf{v}_{\mathbf{k} n} \frac{\partial f^0_{\mathbf{k}n}}{\partial \epsilon_{\mathbf{k}n}} = -\frac{\delta f_{\mathbf{k}n}}{\tau_{\mathbf{k} n}}.$$
于是有: $$\delta f_{\mathbf{k}n} = -q \mathbf{E} \cdot \mathbf{v}_{\mathbf{k} n} \frac{\partial f^0_{\mathbf{k}n}}{\partial \epsilon_{\mathbf{k}n}} \tau_{\mathbf{k} n}.$$
弛豫时间可由($\ref{3}$)式碰撞积分的一般形式计算得到。在准弹性散射近似下,($\omega_{q \lambda}\rightarrow 0$),($\ref{4}$)式的特殊形式成立,($\ref{3}$)式中的碰撞积分可简化为:
$$\left.\frac{\partial f_{\mathbf{k}n}}{\partial t}\right|_{coll}^{RTA} = \sum_{\mathbf{k}'n'} \left(f_{\mathbf{k}'n'}-f_{\mathbf{k}n}\right) P_{\mathbf{k}\mathbf{k'}}^{nn'} \equiv -\frac{\delta f_{\mathbf{k}n}}{\tau_{\mathbf{k} n}}$$,
因为在该近似下,$P_{\mathbf{k}\mathbf{k'}}^{nn'} = P_{\mathbf{k}'\mathbf{k}}^{n'n}$。
由此,我们得到散射几率(弛豫时间倒数)的表达式: $$\frac{1}{\tau_{\mathbf{k} n}} = \sum_{\mathbf{k}'n'} \left(1-\frac{\delta f_{\mathbf{k}'n'}}{\delta f_{\mathbf{k}n}}\right) P_{\mathbf{k}\mathbf{k'}}^{nn'} \tag{5}\label{5}$$
这里,应用了细致平衡方程 $P_{\mathbf{k}\mathbf{k'}}^{nn'}(f^0_{\mathbf{k}'n'}-f^0_{\mathbf{k}n})=0$,并假设散射各向同性。
为计算弛豫时间,我们引入散射角的概念 $$cos(\theta_{\mathbf{k}\mathbf{k'}}) = \frac{\mathbf{v}_{\mathbf{k}'n'} \cdot \mathbf{v}_{\mathbf{k}n}}{|\mathbf{v}_{\mathbf{k}'n'}| |\mathbf{v}_{\mathbf{k}n}|}$$
其中,$\mathbf{v}_{\mathbf{k}n}$ 表示电子速度。由文献【GMSB16】可得: $$\frac{1}{\tau_{\mathbf{k} n}} =\sum_{\mathbf{k}'n'} \frac{(1-f^0_{\mathbf{k}'n'})}{(1-f^0_{\mathbf{k}n})}\left(1-cos(\theta_{\mathbf{k}\mathbf{k'}})\right) P_{\mathbf{k}\mathbf{k'}}^{nn'}$$
迁移率
由弛豫时间,我们可以计算迁移率: $$\mu = -2q \frac{\sum_{\mathbf{k}n} v_{\mathbf{k} n}^2 \frac{\partial f^0_{\mathbf{k}n}}{\partial \epsilon_{\mathbf{k}n}} \tau_{\mathbf{k} n}}{\sum_{\mathbf{k}n}f^0_{\mathbf{k}n}} \tag{6}\label{6}$$
其中因子 2 表示自旋简并。
简化表达式和形变势
由上述公式推导可以看到,迁移率计算是一项大型数值计算任务,这也是采用多个近似的进行简化处理的原因。一个非常常用的近似是形变势近似。电声子耦合矩阵元用解析公式表示,从而直接计算上述公式的积分值。
在形变势近似中,电子-声子耦合项表示为: $$ | \langle \mathbf{k + q}|\delta V_\mathbf{q} | \mathbf{k} \rangle| = |M_{\mathbf{k},\mathbf{q}}| = D^{(1)}\cdot q $$
其中 $D^{(1)}$ 表示一阶形变势。一些声子模式(特别是光学模)零阶形变势已足够准确,即 $|M_{\mathbf{k},\mathbf{q}}|=D^{(0)}$ 与 $q$ 无关。
考虑仅有一个有效声速为$v_{eff}$的声学声子模,产生各向同性一阶形变势$D_{eff}$,迁移率在高温区的解析表达式可写为: $$\mu = \frac{4e\,\hbar\,\rho \,v_F^2 v_{eff}^2}{\pi\, n \, D_{eff}^2 k_B T} \tag{7}\label{7}$$
其中 $\rho$ 表示质量密度,$n$ 为载流子浓度。石墨烯中的 $n$ 与费米能量 $E_F$ 相关,$E_F = \alpha \sqrt{n}$。在高温区,迁移率与温度成反比。在低温极限下,迁移率服从 Bloch-Gruneisen 规律。在 Bloch-Gruneisen 温度 ($T_{BG}=2\hbar\,k_F\,v_s\,/k_B$) 以下,由于反散射 (back-scattering) 的相空间缩减,载流子寿命随温度降低而迅速增加,迁移率比 $T^{-1}$ 更快的增加。【文献KTJ12b】
参考文献
- [GMSB16] (1, 2, 3) T. Gunst, T. Markussen, K. Stokbro, and M. Brandbyge. First-principles method for electron-phonon coupling and electron mobility: Applications to two-dimensional materials. Phys. Rev. B, 93:035414, Jan 2016. doi:10.1103/PhysRevB.93.035414.
- [HS08] EH Hwang and S Das Sarma. Acoustic phonon scattering limited carrier mobility in two-dimensional extrinsic graphene. Physical Review B, 77(11):115449, 2008.
- [KTJ12] K. Kaasbjerg, K. S. Thygesen, and K. W. Jacobsen. Phonon-limited mobility in n-type single-layer mos2 from first principles. Phys. Rev. B, 85:115317, Mar 2012. doi:10.1103/PhysRevB.85.115317.
- [KTJ12b] (1, 2, 3, 4, 5, 6) Kristen Kaasbjerg, Kristian S Thygesen, and Karsten W Jacobsen. Unraveling the acoustic electron-phonon interaction in graphene. Physical Review B, 85(16):165440, 2012.
- [KS92] T Kawamura and S Das Sarma. Phonon-scattering-limited electron mobilities in al x ga 1- x as/gaas heterojunctions. Physical review B, 45(7):3612, 1992.
(本文翻译:朱元慧)