QuantumATK并行计算
概述
如何设置并行计算才能发挥硬件与软件的最佳性能是所有计算程序都要面临的一个问题,这里简单介绍一下并行的的基本概念、机理和设置计算时的一般规律。
基本概念
多核、多路和多节点 现代处理器都是多核处理器,每个处理器包含多个计算处理单元(核),每个单机(节点)又可以包含多个物理处理器,因此概念变得有些模糊。为了清晰器件,我们使用三个层次的结构概念:核、路、节点。
- 节点(node),也叫主机(host)、计算机、机器。一个节点就是一台带有独立物理内存的机器,节点之间通过网络连接,每个节点有确定的IP地址或主机名。
- 路(socket)是指一组通过一个线路与内存相连的计算核心。最常见的情况是一路对应于一个物理的处理器,但这取决于内存和处理器的架构、一路计算核心通常公用L2缓存。
- 核(core)是指一个可以独立进行计算的单元,每个核自有L1缓存。
并行技术
QuantumATK 的并行化包括两个级别:
- MPI (Message Passing Interface)。MPI并行会为每个并行创建独立的内存占用。通常用于ATK-DFT、ATK-SE、ATK-FHI-aims等。
- OpenMP 多线程计算,用于 ATK-Classical 等计算。
QuantumATK 中提到的串行运行指的是没有 MPI 并行,但可以有 OpenMP 多线程计算。MPI 和 OpenMP 的并行方案完全不同,使用这两种并行方案可以大大加速计算。
软件许可(License)需求
QuantumATK 并行计算仅在进行 MPI 多进程并行时才需要并行的 license,在多核上使用 OpenMP 多线程计算不需要额外的 license。
提交一个 QuantumATK 并行计算任务需要一个 master license,用于在启动 MPI 主进程(master process)计算,每增加一个辅助进程(slave process)需要一个 slave license。因此,如果需要提交一个 4 个 MPI 进程的并行计算,需要 1 个 master license 和 3 个 slave license。
通常并行计算需要浮动 license 服务器支持(需要测试并行计算,请单独与我们联系:sales@fermitech.com.cn)。
计算速度的不断提升
此外随着 QuantumATK 软件不断的发展,计算效率也有明显的提升,例如对于这个例子中 4 个 MPI 进程并行计算,ATK2015 与之前版本的比较结果:
| ATK 2015 | ATK 2014 | ATK 13.8 | |
|---|---|---|---|
| Total time | 16m25s | 25m12s | 54m 12s |
| SCF steps | 82/37 | 74/36 | 85/45 |
| I (nA) V=-0.1 V | -6.85 | -6.86 | -6.83 |
- 以上计算体系为 66 原子异质界面器件,在笔记本电脑(Intel i7-3630QM,2.4GHz)上进行 4 MPI 进程的并行计算;使用 SZP 基组,k 点为 1×100,计算内容为分别在 0V 和 -0.1V 偏压下计算透射谱。
综合考虑并行计算性能
MPI 并行计算
对于多数计算而言,使用 MPI 并行可以大大加速计算,因此是推荐选项。 CPU 单线程计算速度主要由 CPU 本身的特性(主频和缓存)决定。MPI 并行效率可以由加速比(计算进程数倍增时获得的加速)来表征,并行效率高低则主要取决于算法本身的特征。 MPI 并行通常将计算拆解为多个独立的“单元计算”,以减少进程间的数据交换。 MPI 并行时,一个计算作业有一个主进程(master)和若干子辅助进程(slave)组成,这也和 QuantumATK 的 license 中的 master 和 slave 对应。Master 和 slave 进程都参与处理单元计算,master 进程还负责进程间的数据传递。
内存消耗
程序单进程串行时,内存消耗主要由计算体系的大小和算法复杂度决定。体系的原子数、电子数以及使用的基组大小,直接决定了矩阵的大小。
单进程运行时需要的内存大小峰值可以在计算之前在 VNL 图形界面上估计。
MPI 并行时,需要为每个 MPI 进程创建独立的内存,例如单进程运行时需要内存为 2G,则在一个节点上进行 24 进程并行时需要 48G 内存。但如果将 24 个 MPI 进程分散在两个节点上,则每个节点需要 24G 内存。
如何在计算前预估内存消耗,请参考:计算对内存的需求。
openMP多线程并行
QuantumATK 可以通过 Intel MKL 使用多线程(openMP)的并行计算,尽管单独使用多线程计算并行效率不如 MPI,但在无 MPI 时仍然可以得到显著的加速。多线程并行的另外一个好处是内存消耗可以明显减小,这是因为多线程并行是共享内存模式的。
MPI+openMP
QuantumATK 中可以实用 MPI 和 openMP 两个级别的并行。目前对于 ATK-DFT 来说应使用 MPI 并行,对于 ATK-Classical 来说使用 openMP 并行。通常不要混合使用 OpenMP 多线程和 MPI 多进程并行。一般来说,除了 ATK-Classical 之外,其他 QuantumATK 计算的 OpenMP 加速有限,因此,强烈建议用户主要使用 MPI 并行,并关闭多线程,即通过设置计算中的环境变量实现:
MKL_NUM_THREADS=1 MKL_DYNAMIC=FALSE
QuantumATK 从 2017 版开始可以方便的使用 MPI+openMP 混合并行,这可以进一步提升并行效率,减少内存消耗。
CPU 时间和内存的矛盾
实际计算时,经常遇到 CPU 时间和内存的矛盾。这是由于计算中需要重复使用的中间数据如果保存在内存里,则可以节约 CPU 时间,加速计算,但将大大增加内存的占用;如果不保存这些中间数据,则可以节约内存,但每次需要这些数据时都要占用 CPU 时间重新进行计算,这将减慢计算速度。
并行效率
并行效率通常以 MPI 并行度增加获得的提速效果来衡量,由于 QuantumATK 中涉及的 DFT 计算(尤其是器件的 DFT 计算,即 DFT-SCF-NEGF)有别于其他DFT 程序的算法,因此横向比较并行效率通常意义不大。目前还缺少针对不同的体系进行完善的并行效率测试结果。针对部分方法测试的结果显示了很好的加速比:
并行的目标
在考虑部署并行计算规模时,需要权衡以下两个目标:
- 在最短的时间内获得计算结果。在一定的范围里不断的增加并行度,通常可以不断的缩短计算时间。
- 并行的经济性。
ATK-DFT的并行
对于常规的ATK-DFT计算,优先使用k点并行,效率较高。通常情况下,QuantumATK根据使用的核心数自动确定并行策略。在并行策略无法达到最优时,QuantumATK会在输出中给出警告(Warning)。此时要达到最有可能需要手动指定核心数使能与k点数整除。
要注意的是,自动选择的并行策略并未考虑内存限制,用户需要自行估计并确定计算的内存消耗状况。
块体材料计算
并行计算基本单元
块体材料的自洽场计算时的基本并行单元计算是k点。 QuantumATK在计算中会使用用户指定的k点布点方案产生k点,并自动判断布里渊区的对称性而得到的不可约的k点,在VNL scripter上会有提示并可以详细查看。

上图显示这个计算有243个独立的k点,因此可以使用大规模并行(这里未考虑内存的限制)。
负载均衡与计算核心数的选择
在选择MPI进程数(核心数)时,应考虑基本单元计算在核心间的负载均衡。例如下图,一个块体计算有两个k点,程序默认尽量将每个k点分配在一个核心上。如果使用3个核心并行,则有一个核心会处于空闲状态;同理,如果使用4核心并行,则程序会选择使用每2个核心处理一个k点;如果使用5个核心,则也会有一个核心处于空闲状态。

“.”表示核心用于计算;“-”表示核心处于空闲。
处理每个k点的进程数
当有多个进程处理一个k点时(例如上例中MPI进程数为4、6、8等时),进程之间的并行由程序自动选择,当然用户也可以指定(processes_per_kpoint)。此参数用于求解将哈密顿量直接对角化得到密度矩阵的DiagonalizationSolver。例如上图中蓝色、红色和绿色的并行策略中使用的process_per_kpoint 分别为 1、2、3。
process_per_kpoint = Automatic 是默认设置,但也可以如下图设置自定义的数值。

举例:
- 在Builder里添加Silver,并进行2x2x2重复;
- 送入Script Generator,双击添加New Calculator,选择ATK-DFT;打开参数设置窗口,选择Algorithm Parameters,点击Parameters:
- 将processes per kpoint设为2,之后用Editor查看:

DiagonalizationSolver的另外一个参数是bands_above_fermi_level,默认设置是All。用户可以自定义该参数,但不能太小,否则会影响对角化程序的精确度。
- 最后,可以在Editor中查看设置的processes_per_kpoint。如下。
# -*- coding: utf-8 -*- # ------------------------------------------------------------- # Bulk Configuration # ------------------------------------------------------------- # Set up lattice lattice = FaceCenteredCubic(8.1714*Angstrom) # Define elements elements = [Silver, Silver, Silver, Silver, Silver, Silver, Silver, Silver] # Define coordinates fractional_coordinates = [[ 0. , 0. , 0. ], [ 0. , -0. , 0.5], [ 0. , 0.5, -0. ], [ 0. , 0.5, 0.5], [ 0.5, 0. , -0. ], [ 0.5, 0. , 0.5], [ 0.5, 0.5, -0. ], [ 0.5, 0.5, 0.5]] # Set up configuration bulk_configuration = BulkConfiguration( bravais_lattice=lattice, elements=elements, fractional_coordinates=fractional_coordinates ) # ------------------------------------------------------------- # Calculator # ------------------------------------------------------------- k_point_sampling = MonkhorstPackGrid( na=3, nb=3, nc=3, ) numerical_accuracy_parameters = NumericalAccuracyParameters( k_point_sampling=k_point_sampling, ) density_matrix_method = DiagonalizationSolver( processes_per_kpoint=2, ) algorithm_parameters = AlgorithmParameters( density_matrix_method=density_matrix_method, ) calculator = LCAOCalculator( numerical_accuracy_parameters=numerical_accuracy_parameters, algorithm_parameters=algorithm_parameters, ) bulk_configuration.setCalculator(calculator) nlprint(bulk_configuration) bulk_configuration.update() nlsave('Silver.nc', bulk_configuration)
注意其中的DiagonalizationSolver的参数process_per_kpoint=2的设置。这个设置最终用于LCAOCalculator的计算。 尝试在2核上运行计算:

或命令行:
$ mpiexec -n 2 atkpython Silver.py > Silver.log
在输出log文件可以看到清楚的表明,两个核心用来处理一个k点,由于只有两个核心,因此只有一个核心组。
+------------------------------------------------------------------------------+ | DiagonalizationSolver parallelization report. | +------------------------------------------------------------------------------+ | Total number of processes: 2 | | Total number of k-points: 14 | | Processes per k-point: 2 | | Number of process groups: 1 | +------------------------------------------------------------------------------+
并行加速与内存消耗峰值
下图显示了ATK-DFT计算的时间和内存消耗峰值可以由于MPI并行而减小。体系有4个不可约的k点。 图中蓝色三角表明了计算时间,红色圆点则代表每个核心的内存消耗的峰值。蓝色水平线表示无并行时(1MPI进程时)的计算时间的100%、50%、25%、12.5%,也就是1核、2核、4核、8核MPI并行的最佳效率。
图中清楚的显示2-4MPI进程并行时获得的加速比最佳,例如使用4核并行时计算时间几乎只有1/4。5-7核并行时则没有明显加速,这是因为增加的核心基本上处于空闲状态。8MPI进程并行时则有明显的加速,此时process_per_kpoint从1变为了2,因此8核全部在工作。 因此,图中用绿色圈出的点就代表了最佳并行效率的MPI数的选择。
从内存角度考虑,最明显的内存降低出现在2MPI并行时,最小的内存消耗出现在process_per_kpoint大于1的时候,这里时8MPI并行时。要注意分布式并行计算的内存消耗峰值的降低很大程度上依赖于计算的体系,通常情况下,大体系有利于并行计算时内存消耗的降低。

NEGF器件体系计算
对于器件体系(DeviceConfiguration和SurfaceConfiguration)两种计算,由于C方向是无限长非周期体系,而A、B方向则有周期性,因此只有AB方向的k点需要考虑,这与块体材料的情形类似。但C方向则不同。
GreensFunction方法使用分块三对角矩阵求逆的方法计算Green's function和lesser Green's function在每个k点和能量点($\varepsilon$)处的数值。lesser Green's function用于计算体系的全密度矩阵:
$$ \begin{split}\hat{D} = - \frac{1}{\pi} \int \int \mathrm{Im} G^< (\varepsilon,k)\ d\varepsilon\ dk\end{split} $$
器件体系的自洽计算涉及能量的Contour点($N_E$)和横向k点(垂直于电子输运方向的k点)($N_k$)为并行计算的基本单元。因此总的并行计算量为 $N_E \times N_k$。
在QuantumATK中,能量($\varepsilon$)的含义对于平衡态和非平衡态的情况是不同的:
- 平衡态时:($\varepsilon$)是复数能量,格林函数使用复平面围道积分(complex contour integral)获得;
- 非平衡态时:($\varepsilon$)是实数能量,格林函数使用实数围道积分(real contour integral)获得。
在实际计算中,积分计算使用两组离散的k点($N_k = [k_1,\ldots,k_\mathrm{n}]$)和能量点($M_\varepsilon = [\varepsilon_1,\ldots,\varepsilon_\mathrm{m}]$—)进行二重加权求和:
$$ \begin{split}\hat{D} = - \frac{1}{\pi} \sum_{i=1}^{N_k} w_{k,i} \sum_{j=1}^{M_\varepsilon} w_{\varepsilon,j} \mathrm{Im} G^< (\varepsilon_j,k_i)\end{split} $$
由于非平衡态下的格林函数使用实数路径积分,因此需要很多的能量$\varepsilon$点,也就是说可以使用非常多的进程并行。
每个$G^<_{i,j}$矩阵元的求值是整个计算过程中最为耗时的,而且对于i和j来说耗时基本相等,因此此两重求和可以一般的写为一组contour点的单重求和$N_k + M_\varepsilon = [x_1,\ldots,x_\mathrm{n+m}]$, 此时包含了能量点和k点:
$$ \begin{split}\hat{D} = - \frac{1}{\pi} \sum_{i=1}^{N_k + M_\varepsilon} w_{k,\varepsilon,i} \mathrm{Im} G^< (x_i)\end{split} $$ 每个contour点即为NEGF计算的一个并行单元。
Contour点自动分布计算
默认情况下,QuantumATK会尽量将NEGF计算的contour点并行分布在尽可能多的进程上。每个contour点都独立计算,即对$N_k \times M_\varepsilon$个contour点有如下并行规则:
- $N_{MPI} = N_k \times M_\varepsilon$ : 每个进程处理一个contour点,理想的最优分布。
- $N_{MPI} <= N_k \times M_\varepsilon$: ATK will calculate groups of \(N_{MPI}\) processes stepwise. QuantumATK每组计算$N_{MPI}$个点,如果$N_{MPI}$不能整除$N_k \times M_\varepsilon$,则有部分进程空置。
- $N_{MPI} > N_k \times M_\varepsilon$ : 计算速度与理想情况的类似,但每个contour点可以使用更多的内存。
每个contour点的进程数
参数选项processes_per_contour_point可以用于控制用于处理每个并行计算单元的进程数。在器件或表面体系计算时,可以如下设置:

默认的processes_per_contour_point=1可以得到最佳的并行效率,对于超大体系,将processes_per_contour_point设为大于1可以增加每个单元计算的进程可用的内存。例如,有$N_k \times M_\varepsilon$个点和$N_k \times M_\varepsilon$个进程,可能每个进程配的内存只有实际需要的一半。如果设置processes_per_contour_point=2即可马上为每个contour点的分配内存增加一倍,此时,所有$N_k \times M_\varepsilon$个点将被分成两组,一组一组的进行计算。
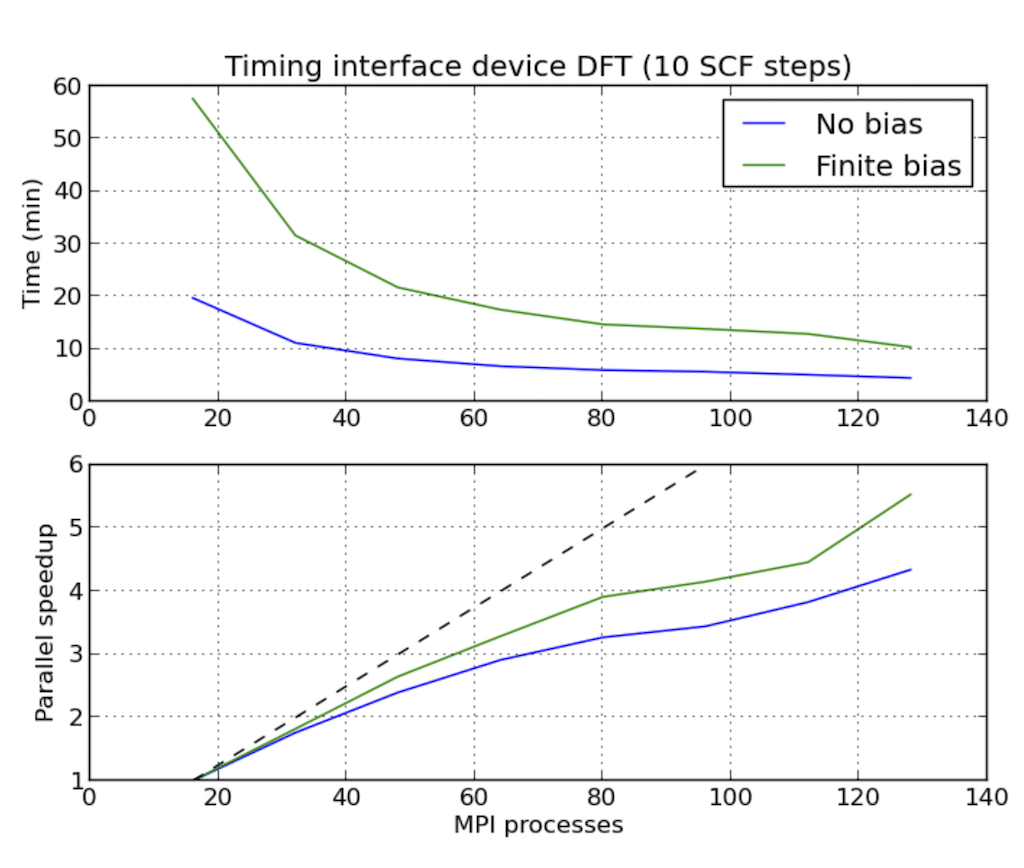
器件计算的并行加速
下图显示了Ag/Si(100)界面体系NEGF计算的并行效率,与文献1)类似。体系计算使用了二维不可约布里渊区的$N_k=10$个k点和$M_\varepsilon$=48个contour点来计算平衡态格林函数。总计$N_k \times M_\varepsilon\$个contour点。

进程数$N_{MPI}$选定为总可以整除480,以避免进程空闲。图中可以看出,并行加速对于测试的进程数都接近理想。processes_per_contour_point = 2 和 processes_per_contour_point = 4 时,计算时间差不多是 processes_per_contour_point = 1 时的二倍或四倍。因为在processes_per_contour_point = 2时,480个点被分成两组240个点先后进行计算。processes_per_contour_point = 4时,480个点被分成四组120个点先后进行计算。但此时,每个点计算的可用内存则为processes_per_contour_point = 1时的二倍或四倍。
QuantumATK中的多级并行
上文介绍了块体材料和器件的单个自洽计算的MPI并行机制。QuantumATK还支持高级的多级并行机制,也就是将复合的计算分解为多个独立的计算在MPI进程上并行,每个独立的计算还可以进一步使用分配给它的MPI进程并行计算。
这类的复合型计算目前包括:IV曲线计算、NEB过渡态搜索计算、adaptive kinetic Monte Carlo(AKMC)计算、晶体结构预测(基因算法全局优化)计算。
所有这些计算都涉及同时对多个结构进行独立的计算,多级并行可以同时进行这些计算,因此可以明显缩短得到计算结果的时间。
多级别的并行是自动启用的,当然也可以在VNL Scripter – New Calculator – Parallelization parameter中进行设置。

IV计算
IV曲线计算是在对器件体系进行多个偏压点的NEGF计算,因此可以对偏压点和NEGF计算进行两个级别并行(如图):
- 并行级别I.192个核心被分为两组,每组96核心,分别处理偏压#1和偏压#2。QuantumATK可以自动选择每个偏压点使用多少核心,也可以手动设置。
- 并行级别II.每组核心(96核)可以进一步并行处理contour点(1个能量的1个k点),如果体系是48能量点*2k点,则96核心刚好全部用上。每个contour点使用几个核心可以由QuantumATK自动选择或手动设置。

NEB计算
如果使用64核心(64MPI进程)对一个NEB计算(共4个结构,每个结构2个不可约k点)进行和多级并行。下图显示了多级别并行的机制。
- 并行级别I. 64核心分成四组(每组16核心)分别处理四个NEB结构。(每个NEB结构用几个MPI进程处理可以通过VNL Scripter或脚本中设置)
- 并行级别II.每组的16个核心又可以分为2组,每组8核心处理一个k点。QuantumATK可以自动选择并行数,当然,也可以在New Calculator → Algorithm Parameters → DiagonalizationSolver Parameters中设置。
- 并行级别III.QuantumATK自动设置这一级别并行。

AKMC计算
AKMC特别适合多级别并行,因为计算时需要进行多个鞍点的搜索。QuantumATK默认会尽可能的在核心上均分作业负载。例如使用ATK-DFT进行并行时:
- 192核心被分成48组,每四个核心一组用于处理一个鞍点。QuantumATK默认会自动进行这种分组,当然也可以进行手动设置。
- 每组的4个核心可以进一步分开并行处理每个k点。例如体系有2个k点,则每个k点由2个核心处理。
- 下一级别的并行由QuantumATK自动选择。

晶体结构预测计算
全局优化计算使用多代晶体结构演化的算法来搜索最低能量结构,每一代都有几百甚至上千结构,因此可以高效的使用多级并行计算:
- 192核心分成48组,每4个核心一组用于处理一个构型。分组可以由QuantumATK自动完成,也可以手动设置。
- 当使用DFT进行计算时,每组的4个核心可以用于进行k点并行。
- 下一级别的并行由QuantumATK自动选择。
